Phylogenetik- Veränderung in Genomen
Assistenzprofessor Dr. Shigehiro Kuraku,
Labor für Zoologie und Evolutionsbiologie,
Fachbereich Biologie, Universität Konstanz
Es war der Traum der Biologen den Mechanismus zu entschlüsseln, der die Entwicklung der unterschiedlichen Erscheinungsformen der Organismen dieses Planeten erlaubt hat. Die Etablierung der evolutionären Entwicklungsbiologie hat eine Hypothese geliefert,
dass viele regulatorische Gene, die an der Entwicklung selbst entfernt verwandter
Tiere beteiligt sind, identisch sind. Die seit kurzem im Großmaßstab verfügbaren DNA-Sequenzressourcen bieten die Möglichkeit diese Idee durch genomweite Analysen zu untersuchen. Die Brücke zu schlagen zwischen morphologischer Diversität und molekularen Merkmalen ist eine herausfordernde Aufgabe. Diese Grenze scheint nur durch eine Kombination verschiedener biologischer Ansätze erreichbar, die zu einer integrativen, evolutionären Studie in der post-genomischen Ära beitragen.

Kyoto, Kobe und Konstanz Kyoto, eine historische Stadt im Westen Japans, ist der
Platz an dem ich, in der Startphase meiner wissenschaftlichen Laufbahn, davon geträumt habe eines Tages Wissenschaftler in Übersee zu werden. Später bin ich in eine moderne Stadt umgezogen, Kobe, wahrscheinlich am bekanntesten durch das starke Erdbeben von 1995.
Im Frühjahr 2007 bin ich nach Konstanz gekommen, mit herrlichem Blick auf den Boden- see. Ein 10-Minuten-Spaziergang von der Stadtmitte führt einen in die Schweiz – deshalb ist Konstanz ein Ort der grenzenlosen Gedanken. Letztes Jahr wurde Konstanz als eine der 9 Exzellenz-Universitäten Deutschlands ausgewählt. Bevor ich nach Deutschland kam, konnte ich mir eine derartige (Be-)Förderung meines nächsten Arbeitsplatzes nicht vor- stellen, was natürlich ein großes Plus darstellt. Ob es eine Standardsichtweise ist oder nicht, mir wurde von meinem letzten Betreuer eingeprägt, dass Deutschland ein Land der evolutionären Morphologie ist, geprägt durch Johann Wolfgang von Goethe, Karl Ernst von Baer, Ernst Haeckel … Im Gegensatz dazu blickt Japan eher auf eine Geschichte der evolutionären Untersuchungen auf molekularer Ebene zurück. Es gibt hier viele berühmte
Meilensteine, wie sich DNA-Sequenzen entwickelt haben (nämlich die der molekularen Evolution). Der hervorstechendste ist der der „neutralen Theorie der molekularen Evolution“, bereits vor etwa 40 Jahren von Motoo Kimura vorgeschlagen [1]. Diese Theorie schlägt vor, dass sich DNA-Sequenzen über die Zeit mit neutralen Mutationen ändern und ist bekannt durch den berühmten Satz „Überleben des Glücklichsten“ statt „Überleben des Fittesten“,
gemünzt auf die Übertragung des Konzeptes der natürlichen Selektion der morphologischen Evolution, begründet durch Charles Darwin, dessen 200. Geburtstag dieses Jahr gefeiert wird. Selbst bis heute ist der Gegensatz zwischen diesen beiden Ebenen der Evolution – morphologischer und molekularer – noch nicht in Einklang gebracht.
Evolutionäre Neuheiten
Klar, Studien zur morphologischen Evolution gingen denen der DNA-Sequenzen voraus. Als typische Abfolge der Ableitung in der morphologischen Evolution erkennen wir eine charakteristische morphologische Struktur eigens für Mitglieder einer bestimmten Gruppe von Tieren und schließen daraus, dass der Vorfahr dieser Gruppe dieses Charakteristikum erworben hat. Ein Beispiel:
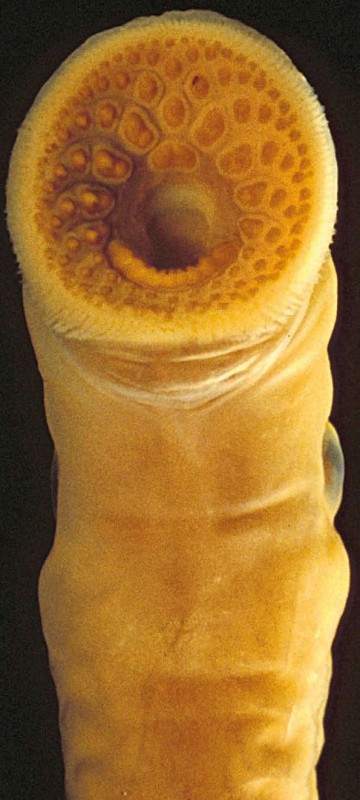
Alle lebenden Schildkröten haben einen Panzer auf dem Rücken (Abb. 1A). Während der Schildkröten-Embryonalentwicklung im Ei-Chorion haben alle einen rudimentären Panzer. Ich schlage in einer meiner früheren Studien vor, dass Änderungen im Expressionsmuster universell vorhandener Gene diverser Wirbeltiere diesen großen Sprung im Körper-Bauplan verursacht haben [2, 3] – morphologische Evolution kann ohne das Hinzukommen neuer Gene auftreten. Eine andere fundamentale Neuheit in der Wirbeltier-Evolution, der Kiefer (Abb. 1B), untersuchen wir durch Vergleich von Tieren mit und ohne Kiefer (Abb. 2) [4]. Offensichtlich erfordert diese Studienrichtung breites biologisches Wissen, verfügbar in
Form von Beispielen aus der Tierwelt (üblicherweise nicht Modellorganismen) und geeignete experimentelle Werkzeuge.

Baum-Denken
Um evolutionäre Ereignisse in phylogenetischen Bäumen zu kartieren, wie in Abb. 1, müssen wir im Voraus die Beziehung zwischen den fraglichen Tieren kennen.
Heute können Forscher DNA-Sequenzen verschiedener Tiere im Großmaßstab nutzen – Hunderte von Genen oder mehr – um die entsprechenden Regionen untereinander zu vergleichen und dadurch den molekularen phylogenetischen Baum zu rekonstruieren. Die Anwendung molekularer phylogenetischer Bäume ist nicht nur auf Evolutionsstudien begrenzt. Man kann molekulare phylogenetische Bäume zur Charakterisierung neu entdeckter Gene verwenden und dann auf ihren Ursprung und ihre Funktion durch Abgleich mit den Eigenschaften des phylogenetischen Nachbarn rückschließen. In diesem Sinne ist molekulare Phylogenetik ein unersetzbarer Teil der Molekularbiologie. Aufgrund eines Mechanismus, der eine Kopie eines existierenden Genes anfertigt, Gen-Duplikation genannt, kann ein einzelnes Genom vielfache Versionen ähnlicher Gene enthalten. Solche Verwandte werden in einer Genfamilie kategorisiert. Weil ein einzelnes Genom viele Mitglieder von Hunderten von Genfamilien enthält, erhalten wir bei Ableitung molekularer phylogenetischer Bäume für individuelle Genfamilien einen Überblick über die Geschichte der Genomevolution, die zum Teil durch Gen-Duplikation geformt wurde. Gleichzeitig gehen existierende Gene häufig während der Evolution verloren. Ein interessantes Beispiel dafür
ist das „homeobox-containing“ Gen Hox14, das im Neunauge, dem Hai, den Quasten-flossern und Lungenfischen, nicht aber in den üblicherweise im Labor verwendeten
Organismen gefunden wird, wie Maus, Huhn und Zebrafisch (Abb. 3). Da die Erhaltung eines Genes im Genom ein Zeichen für seine Funktionalität ist, Entspannung solcher funktioneller Zwänge kann die sekundären Verluste der Hox14 Gene erlaubt haben [5]. Wichtig anzumerken, Tiergruppen mit diesem Gen sind nicht notwendigerweise ühylogenetisch nah verwandt. Der evolutionäre Prozess ist viel komplizierter als eine einfache Serie von uni-direktionalen Additionen neuer Komponenten. Daher sind wir auch heute auf baumbasierte Ansätze angewiesen, um ausführlich die Geschichte der Genom-
Evolution zu beschreiben. Da Tausende oder mehr Gene pro Genom neu identifiziert werden, steigt die Wichtigkeit der molekularen Phylogenetik zusammen mit dem
aufkommenden Feld der Bioinformatik.

Sind wir voreingenommen?
Wirbeltiere werden durch viele hoch entwickelte, phenotypische Eigenschaften definiert, so wie die Neuralleiste und, wie der Name impliziert, der Wirbel. Die Etablierung dieser fortgeschrittenen Eigenschaften gilt als die finale große Verbesserung auf dem langen evolutionären Weg vom Bakterium zu uns Menschen. Zurzeit konzentriert sich die Sequenzierung von Wirbeltier-Genomen allerdings nur auf Wirbeltiere mit Knochen. Unglücklicherweise werden früh abgezweigte Wirbeltiere (z.B. Neunaugen, Schleimaale, Haie, Rochen, Chimären) nicht berücksichtigt (Abb. 3). Das rührt zum Teil daher, dass
diese Tiere für intensive Erforschung schwer im Labor zu halten sind. Neunaugen-Embryos zum Beispiel waren bis vor kurzem für mehr als 100 Jahre nicht verfügbar [6].
Unglücklicherweise kann man nicht begründen „was Wirbeltiere sind“ ohne diese früh abgezweigten Tiere untersucht zu haben.
Baum-Verrückte für Genomanalyse*
Ich gehe leidenschaftlich gern auf die Jagd nach Antworten zu generellen Fragen in der Biologie durch molekulare Phylogenetik. Zurzeit ist eines meiner Ziele den Zeitpunkt der Gesamtgenom-Duplikation in der frühen Wirbeltier-Evolution herauszufinden. Ist es möglich, dass dieses Ereignis, zuerst vorgeschlagen vor ungefähr 40 Jahren von dem bekannten japanischen Forscher Susumu Ohno [7], die Hauptquelle für die endgültige Verbesserung
der Genom-Struktur ist, die zum menschlichen Genom geführt hat [8,9]. Einige meiner Mitarbeiter arbeiten auch an anderen Projekten, um die Beziehung zwischen Genregulation und Genlokation in Genomen herauszufinden und um Genomeigenschaften zu identifizieren,
die die schnelle Diversifizierung von Arten verursachen.
Die aufkommenden im Großmaßstab verfügbaren DNASequenzdaten haben Möglichkeiten geschaffen lange offene Fragen der Evolution zu lösen. Es besteht kein Zweifel daran, dass uns Evo-Devo-Konzepte und molekulare, phylogenetische Methoden, ebenso bio- informatische Ansätze helfen werden, die grenzenlosen Gedanken in die Praxis umzusetzen.
shigehiro.kuraku@uni-konstanz.de
Der Beitrag ist eine Übersetzung des englischen Originalartikels von Shigehiro Kuraku
„Phylogenomic Evo-Devo - In genomes, what can change and what cannot?“,
der in der internationalen Ausgabe lab&more0109 veröffentlicht wird.
Foto: © Dr. Shigehiro Kuraku
Literatur
[1] Kimura M: Evolutionary rate at the molecular level. Nature 1968,
217(5129):624-626.
[2] Kuraku S, Usuda R, Kuratani S: Comprehensive survey of carapacial ridgespecific
genes in turtle implies co-option of some regulatory genes in carapace
evolution. Evol Dev 2005, 7(1):3-17.
[3] Nagashima H, Kuraku S, Uchida K, Ohya YK, Narita Y, Kuratani S: On the
carapacial ridge in turtle embryos: its developmental origin, function and the
chelonian body plan. Development 2007, 134(12):2219-2226.
[4] Takio Y, Pasqualetti M, Kuraku S, Hirano S, Rijli FM, Kuratani S: Evolutionary
biology: lamprey Hox genes and the evolution of jaws. Nature 2004,
429(6989):1 p following 262.
[5] Kuraku S, Takio Y, Tamura K, Aono H, Meyer A, Kuratani S: Noncanonical
role of Hox14 revealed by its expression patterns in lamprey and shark.
Proc Natl Acad Sci U S A 2008, 105(18):6679-6683.
[6] Ota KG, Kuraku S, Kuratani S: Hagfish embryology with reference to the
evolution of the neural crest. Nature 2007, 446(7136):672-675.
[7] Ohno S: Evolution by gene duplication. New York: Springer-Verlag; 1970.
[8] Kuraku S: Insights into cyclostome phylogenomics: pre-2R or post-2R.
Zool Sci 2008, 25(10): 960-968.
[9] Kuraku S, Meyer A, Kuratani S: Timing of whole genome duplications relative
to the origin of the vertebrates: did cyclostomes
Stichwörter:
Phylogenetik, Genom, Evolution
|
L&M 1 / 2009
Diese Artikel wurden veröffentlicht in Ausgabe L&M 1 / 2009.
Das komplette Heft zum kostenlosen Download finden Sie hier:
zum Download
Der Autor:
Weitere Artikel online lesen
News
Mit dem HPLC-Säulenkonfigurator unter www.analytics-shop.com können Sie stets die passende Säule für jedes Trennproblem finden. Dank innovativer Filtermöglichkeiten können Sie in Sekundenschnelle nach gewünschtem Durchmesser, Länge, Porengröße, Säulenbezeichnung u.v.m. selektieren. So erhalten Sie aus über 70.000 verschiedenen HPLC-Säulen das passende Ergebnis für Ihre Anwendung und können zwischen allen gängigen Herstellern wie Agilent, Waters, ThermoScientific, Merck, Sigma-Aldrich, Chiral, Macherey-Nagel u.v.a. wählen. Ergänzend stehen Ihnen die HPLC-Experten von Altmann Analytik beratend zur Seite – testen Sie jetzt den kostenlosen HPLC-Säulenkonfigurator!
© Text und Bild: Altmann Analytik
Aufnahme, Dokumentation und Teilen von Ergebnissen mit ZEISS Stemi 305 und ZEISS Stemi 508
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen.
© Text und Bild: Carl Zeiss Microscopy GmbH
|