


|
L&M-3-2011
>
Therapie der seltenen Erkrankung alpha-Mannosidose
Therapie der seltenen Erkrankung alpha-MannosidoseEuropäische Union ermöglicht TherapieFür seltene Erkrankungen besteht meist wenig Aussicht auf Heilung, da insbesondere das wirtschaftliche Interesse der Pharmafirmen zu gering ist, in die Entwicklung effektiver Therapien zu investieren. Ohne öffentliche Förderung besteht somit wenig Aussicht, seltene Erkrankungen erfolgreich zu behandeln. Die lysosomale Speichererkrankung alpha-Mannosidose, die weltweit weniger als 500 Menschen betrifft, ist eine solche seltene Erkrankung. Sie betrifft meist Kinder und führt unbehandelt zum Tode der Betroffenen. Eine früh eingesetzte Therapie könnte aber den fatalen Verlauf der Krankheit verhindern. Durch die langjährige Förderung der Europäischen Union und die Initiative von Wissenschaftlern und Ärzten aus ganz Europa ist es nun gelungen, eine Therapie für die alpha-Mannosidose zu entwickeln, die derzeit an Patienten klinisch getestet wird. Lysosomale Speichererkrankungen (LSD) Die LSD alpha-Mannosidose ist eine seltene genetische Erkrankung mit einer Verbreitung von etwa 1 zu 500000, die weltweit weniger als 500 Menschen betrifft und deshalb nach Vorgaben der Europäischen Union (EU) als eine „sehr seltene“ Erkrankung eingestuft wird. Aufgrund des geringen Interesses der Pharmafirmen besteht für diese Arten von seltenen Erkrankungen ohne öffentliche Förderung wenig Aussicht auf die Entwicklung einer effektiven Therapie. LSD umfassen eine Gruppe von etwa 50 vererbbaren Stoffwechselerkrankungen, die hauptsächlich durch Mutationen in Genen, die für lysosomale Enzyme kodieren, verursacht werden. Charakteristisch für LSD sind der autosomal rezessive Erbgang, ihr früher Beginn, da meist Kinder betroffen sind, und die häufige Beeinträchtigung des Gehirns, was die Therapie dieser Erkrankungen erschwert. Unbehandelt führen die LSD meist zum Tode der Betroffenen. Lysosomen – ein ausgeklügeltes Recyclingsystem der Zelle Lysosomen sind zytoplasmatische, membranumgebende Organellen, in deren Inneren ein saurer pH-Wert herrscht [4]. Lysosomen finden sich in allen unterschiedlichen Zellen des Körpers. Der saure pH-Wert im Inneren dieses Organells ist für die Aktivität der lysosomalen Enzyme, den so genannten sauren Hydrolasen, wichtig, die Makromoleküle wie z.B. Zucker, Fette und Proteine in ihre Bausteine zerlegen. Diese Bausteine werden dann durch die Aktivität von Transportproteinen in der lysosomalen Membran aus dem Lysosom heraustransportiert und der Zelle für die Synthese neuer Makromoleküle zur Verfügung gestellt [4]. Dieses ausgeklügelte Recyclingsystem der Zelle funktioniert allerdings nur dann, wenn sich der Abbau und der Aufbau dieser Makromoleküle die Waage halten. Kommt es wie bei den LSD zu einer krankhaften Speicherung von Makromolekülen innerhalb des Lysosoms, führt dies langfristig zu einem Zusammenbruch der lysosomalen Funktionen in der Zelle mit fatalen Folgen für den Organismus, was in der Schwere der Krankheitsbilder der LSD zum Ausdruck kommt [2]. Obwohl die Zellen von Geburt an Speichermaterial aufweisen, sind die meisten der betroffenen Kinder bei der Geburt unauffällig. Die ersten Symptome der Krankheit zeigen sich dann – je nach Schweregrad der Erkrankung – im Säuglings-, frühen Kindes- oder Erwachsenenalter und betreffen viele Organsysteme, darunter auch das zentrale Nervensystem. Der Verlauf der LSD ist progredient. alpha-Mannosidose Bei der alpha-Mannosidose unterscheidet man trotz einer großen Heterogenität in Bezug auf das klinische Erscheinungsbild drei Formen (Typ 1–3) [1], die sich vor allem in der Schwere des Krankheitsverlaufs, einer neuronalen Beteiligung und im Beginn der klinischen Symptomatik unterscheiden. Die meisten Patienten gehören der moderaten Form an (Typ 2), die innerhalb der ersten Lebensdekade diagnostiziert wird. Da die betroffenen Kinder „gesund“ auf die Welt kommen, könnte besonders in diesen Fällen eine früh eingesetzte Therapie den fatalen Verlauf der Krankheit verhindern. Hervorgerufen wird die alpha-Mannosidose durch Mutationen im Gen Man2B1, das für die lysosomale saure Hydrolase alpha-Mannosidase (LAMAN) kodiert [1, 5, 6]. LAMAN ist in gesunden Zellen am Abbau von mannosehaltigen Zuckern beteiligt. Bei dem Verlust von LAMAN akkumulieren diese mannosehaltigen Zucker in der Zelle und führen beim Menschen zu einer Immunschwäche, fazialen Anomalien, Skelettveränderungen, Schmerzen, Schwerhörigkeit und intellektuellen Defiziten. Derzeit ist die alpha-Mannosidose nicht heilbar und die Patienten können nur symptomatisch behandelt werden. Der Krankheitsverlauf kann durch diese Behandlungsform nicht aufgehalten werden und die Patienten versterben. Enzymersatztherapie (ERT) Trotz enormer Kosten und gewisser Einschränkungen ist die bislang viel versprechendste Therapie für Patienten mit LSD einschließlich der alpha-annosidose die ERT. Bei dieser Therapieform wird das fehlende Enzym mithilfe gentechnischer Methoden in kultivierten Zellen hergestellt und kann dann in aufgereinigter Form den Patienten intravenös verabreicht werden. Das Enzym zirkuliert nun im Gefäßsystem des Patienten und wird idealerweise von den Zellen verschiedener Gewebe über rezeptorvermittelte Endozytose aufgenommen und nach Aufnahme in die Zelle in das Lysosom transportiert, wo es das fehlende Enzym ersetzt und das akkumulierte Speichermaterial abbaut. Wird diese Therapie zu einem frühen Zeitpunkt eingesetzt, kann durch die Normalisierung der lysosomalen Funktionen der progrediente Verlauf der Erkrankung verhindert werden. Die Patienten sind lebenslänglich auf diese Therapie angewiesen. Es sind bereits einige Enzyme für die Behandlung von LSD auf dem pharmazeutischen Markt erhältlich wie z.B. für Morbus Gaucher, Morbus Fabry, Morbus Pompe und die Mukopolysaccharidosen vom Typ I, II und VI. Mehrere klinische Studien für die Behandlung weiterer LSD sind derzeit am Laufen. Ein Nachteil der ERT besteht allerdings darin, dass angenommen wird, dass rekombinante lysosomale Enzmye die Blut-Hirn-Schranke nicht passieren können, was die Effizienz einer Behandlung von LSD mit neuronaler Beteiligung stark einschränkt. Präklinische ERT an einem Mausmodell für alpha-Mannosidose
Genetisch veränderte Mäuse werden weltweit z.T. sehr erfolgreich als Krankheitsmodelle zur Erforschung und Therapieentwicklung genetischer Erkrankungen eingesetzt wie z.B. Morbus Alzheimer, da an ihnen – im Gegensatz zum Menschen – gewebespezifische Untersuchungen zur Effizienz einer räklinischen Therapie erfolgen können – auch für die alpha-Mannosidose existieren Mausmodelle, die die humane Erkrankung in vielen Aspekten widerspiegeln [7, 8]. Das Speichermaterial lässt sich in Mausgeweben sowohl biochemisch über chromatografische Methoden als auch morphologisch nachweisen (siehe Abb.1–2). Die Erforschung seltener Erkrankung – ein Schwerpunkt der EU Die weltweite Verbreitung von LSD unter Lebendgeborenen wird mit 11–15 Betroffenen pro 100.000 angegeben. Nach der Verordnung der EU wird eine Krankheit als selten bezeichnet, wenn sie weniger als 5 Menschen auf 10.000 betrifft. Fasst man alle seltenen Erkrankungen zusammen, so kommt man auf eine Gruppe von etwa 20 – 30 Mio. Europäern, die von einer seltenen Erkrankung betroffen sind. Da viele der seltenen Erkrankungen lebensbedrohlich bzw. in ihrem Verlauf chronisch sind, haben sie einen dramatischen Einfluss auf die Lebenserwartung und –qualität sowohl der Betroffenen als auch ihrer Familien. Darüber hinaus fallen durch die langjährige intensive medizinische Betreuung der Patienten hohe Kosten im Gesundheitswesen an. Ein großes Problem bei der Behandlung seltener Erkrankungen ist das Fehlen effektiver Therapien. Aufgrund ihrer Seltenheit und des Fehlens lukrativer Absatzmöglichkeiten ist das Interesse von Pharmafirmen gering, Geld für die Entwicklung von Medikamenten in die Behandlung seltener Erkrankungen zu investieren. Deshalb hat die Europäische Union in den letzten Jahren hohe Fördermittel für wissenschaftliche Projekte zur Erforschung und Behandlung seltener Erkrankungen zur Verfügung gestellt. Das europäische Netzwerk ALPHA-MAN
Diese von der EU bereitgestellten Fördermittel wurden von einem europäischen Netzwerk, bestehend aus Wissenschaftlern, Ärzten und mittelständischen biotechnologischen Unternehmen, verwandt, um eine Enzymersatztherapie für die sehr seltene alpha-Mannosidose zu entwickeln. Das erste Netzwerk EURAMAN (2001 – 2005), das unter der Führung von Prof. Dr. Kurt von Figura (Universität Göttingen) im Rahmenprogramm 5 der EU vor 10 Jahren ins Leben gerufen wurde, bildete die Basis, auf der die weiteren Netzwerke unter der Leitung von Prof. Dr. Paul Saftig und Dr. Judith Blanz (Universität Kiel) HUEMAN (2006 – 2009, Rahmenprogramm 6) und ALPHA-MAN (2010 – 2013, Rahmenprogramm 7) aufbauten – alle mit dem Ziel, eine Enzymersatztherapie an alpha-Mannosidose- Patienten möglich zu machen. Innerhalb des EURAMAN und HUE-MAN Konsortiums wurden folgende Ergebnisse erreicht: i) Erfassung aller in Europa auftretenden klinischen MAN2B1-Mutationen und deren biochemische Charakterisierung, ii) Etablierung diagnostischer Tests, iii) Erstellung einer öffentlichen Datenbank (http:/
jblanz@biochem.uni-kiel.de, Literatur
[1] Malm, D. and O. Nilssen, Alpha-mannosidosis. Orphanet J Rare Dis, 2008. 3: p. 21. |
L&M 3 / 2011
Das komplette Heft zum kostenlosen Download finden Sie hier: zum Download Die Autoren:Weitere Artikel online lesen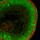



NewsSchnell und einfach die passende Trennsäule finden
© Text und Bild: Altmann Analytik ZEISS stellt neue Stereomikroskope vor
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen. © Text und Bild: Carl Zeiss Microscopy GmbH |






