|
L&M-5-2008
>
ncRNA - nicht codierende regulatorische RNA
ncRNA - nicht codierende regulatorische RNAEin neues Verständnis des humanen Genoms
Prof. Dr. John S. Mattick Institut für Molekulare Biowissenschaft, University of Queensland, Brisbane, Australien 
Das Konzept, dass Gene über das Intermediat messenger RNA (mRNA) für Proteine kodieren, durchzieht die Molekularbiologie seit ihren Ursprüngen, so wie der Gedanke, dass eine steigende Komplexität durch eine größere Zahl an (Protein-kodierenden) Genen unterstützt wird. In der Tat besitzen Prokaryoten, die eine größere metabolische und ökologische Flexibilität zeigen, ein größeres Repertoire an Protein-kodierenden Genen und komplexere Organismen haben mehr Protein-kodierende Gene als Einzeller. Wie auch immer, die angenommene Beziehung zwischen Genzahl und Entwicklungsgrad wird zunehmend angezweifelt, da herausgefunden wurde, dass der einfache Nematode Caenorhabditis elegans mit nur ~ 1.000 Zellen die gleiche Zahl an Protein-kodierenden Genen (~ 20.000) besitzt wie Menschen mit ~ 100 Trillionen Zellen, die präzise in unzähligen, fein-skulpturierten Geweben angeordnet sind, inklusive eines Gehirns mit weitentwickelten kognitiven Funktionen. Darüber hinaus haben viele in den Genomen von C. elegans und Menschen kodierten Proteine orthologe Funktionen. Obwohl das Proteom des Menschen durch die verstärkte Anwendung alternativen Splicings vergrößert wurde und zusätzlich Stamm-spezifische Gene existieren, der Umfang an Protein-kodierenden Sequenzen ändert sich nicht merkbar zwischen Wurm und Mensch, was darauf hinweist, dass die für die Programmierung einer fortgeschritteneren Entwicklungskomplexität erforderliche relevante Information hauptsächlich in den nicht-kodierenden Regionen des Genoms liegen muss, welche im Umfang bei komplexeren Organismen wachsen [1]. Das wiederum lässt den Schluss zu, dass diese nicht-kodierenden Regionen für ein zunehmend ausgeklügelteres regulatorisches System zur Kontrolle der Expression und der Verwendung des Proteoms kodieren, was mit der Beobachtung korreliert, dass wenigstens bei Prokaryoten, wo die meisten regulatorischen Vorgänge von Proteinen durchgeführt werden, das Verhältnis der regulatorischen Gene mit wachsender Genomgröße ansteigt [2].
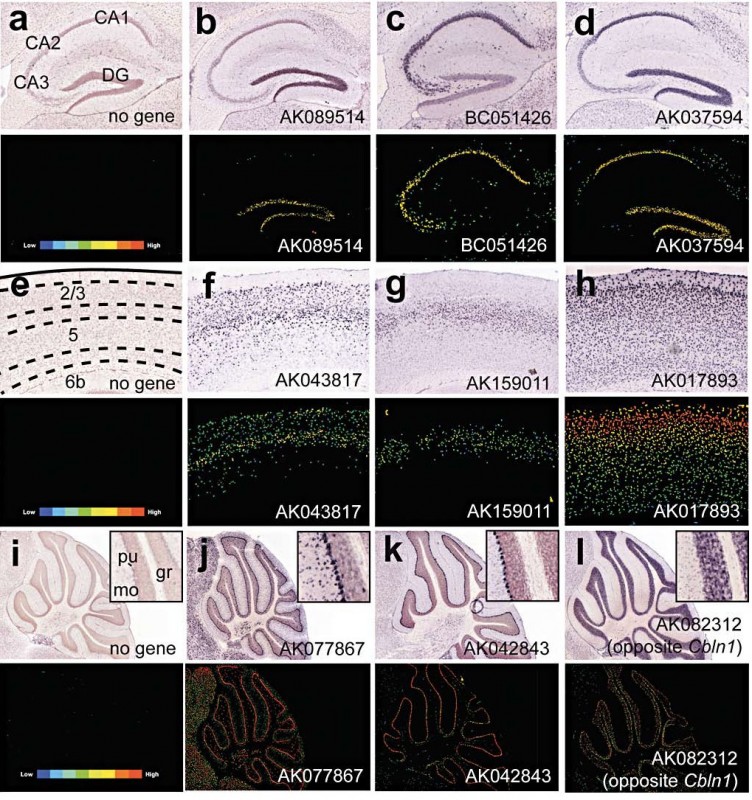
Der Vorschlag, dass die extensiven intronischen und Intergen-nicht-kodierenden Regionen des Genoms der Säuger und anderer komplexer Organismen genetisch aktiv sein mögen, wird durch neuere Erkenntnisse stark gestützt, nämlich dass die überwiegende Mehrheit des Genoms, von Würmern bis Menschen, in einer entwicklungsabhängigen Weise transkribiert wird, hauptsächlich in nicht-Protein-kodierende RNAs (ncRNAs). Diese Transkripte werden in erstaunlich komplexen Mustern von überlappenden und vernetzten Transkripten exprimiert, häufig weite Regionen des Genoms überspannend, was darauf hinweist, dass sie ununterbrochen Information enthalten [3]. Gleichzeitig gibt es zunehmend Hinweise auf hoch entwickelte Netzwerke RNA-basierter regulatorischer Systeme in höheren Eukaryoten. Diese schließen die gut charakterisierten miRNAs und siRNAs ein, die die mRNATranslation und Stabilität kontrollieren (von denen die meisten noch entdeckt werden müssen, speziell die, die in begrenzten Zellpopulationen und/oder Speziesspezifisch exprimiert werden), siRNAs, die das transkriptionelle „gene silencing“ bewirken und Heterochromatin-Bildung kontrollieren und piRNAs, die die Transposon-Aktivität in Keim- und somatischen Zellen von Fliegen bis Menschen kontrollieren. Sie schließen auch ein wachsendes Repertoire an längeren ncRNAs ein, von denen Zehn-, wenn nicht sogar Hunderttausende in Säugern vorhanden sind [3]. Diese ncRNAs werden dynamisch während der Differenzierung und Entwicklung exprimiert [2,4], einschließlich der embryonalen Stammzell-Differenzierung [5] und im Hirn [6]. Sie zeigen präzise zelluläre und subzelluläre Lokalisationen, ein Hinweis auf funktionale Spezifität (Abb. 1).
Solche Beobachtungen stehen im Widerspruch mit der verbreiteten Ansicht, dass nur ~ 5 % des menschlichen Genoms „konserviert“ sind (d. h. unter reinigender Selektion für Funktionen typisch für Säuger) und daher funktional. Wie auch immer, dieses Abbild schließt solche Sequenzen aus, die abstammungsspezifisch sind und solche die ortholog sein mögen, aber relativ schnell abgezweigt sind, im Vergleich zu Protein-kodierenden Sequenzen und einigen regulatorischen Sequenzen, die durch strikte Struktur-Funktions-Beziehungen oder multilaterale Interaktionen eingeschränkt sind. Dies ist auch auf der fraglichen Annahme begründet, dass sehr alte, aus Transposons stammende Sequenzen, die als Referenz-Sequenzen für die Rate „neutraler Evolution“ verwendet werden, nicht funktional sind, trotz der Tatsache, dass sie in unseren Genomen (in einigen Fällen) seit Hunderten von Millionen Jahren ko-existieren. Daher sollte man beim eukaryotischen Genom eher an eine RNA-Maschine denken, als es als Inseln von Protein-kodierenden Genen in einer sich ausweitenden See aus evolutionärem „junk“ zu sehen [10]. Vorausgesetzt, dass Menschen dasselbe Proteom besitzen und dass dieses größtenteils mit dem anderer Säuger und Vertebraten übereinstimmt, dann ist der klare Schluss daraus, dass der Großteil der Unterschiede zwischen Individuen und Spezies wahrscheinlich in der RNA-basierten Kontrollarchitektur eingebettet ist. Der Grund, warum dies nicht früher erkannt wurde, liegt an dem Protein-fokussierten Blick auf die genetische Programmierung, was zunehmend primitiv erscheint, einhergehend mit der Tatsache, dass die meisten Variationen in regulatorischen Sequenzen nur sehr fein sind und bis vor kurzem nicht im Detail untersucht wurden (oder untersucht werden konnten), im Gegensatz zu Mutationen in Protein-kodierenden Sequenzen, die oft desaströse Konsequenzen haben. Wir scheinen an einem Neubeginn in unserem Verständnis der genetischen Programmierung der menschlichen Entwicklung zu stehen, so wie wir anfangen die Funktionen von ncRNA nicht nur beim Spezifizieren unserer Evolution, Ontogenese und individuellen Differenzen zu erforschen, sowohl physisch wie auch mental, sondern auch in den molekularen Ursachen von Krebs und anderen komplexen Krankheiten. Das wird enorme praktische Folgerungen und Konsequenzen haben und wird ein Unternehmen sein, das die molekulare Biologie in absehbarer Zeit einnehmen wird. Wir danken Applied Biosystems für die Erlaubnis die Fotomontage von John Mattick zu verwenden. Foto: © Prof. Dr. John S. Mattick
Literatur |
L&M 5 / 2008
Das komplette Heft zum kostenlosen Download finden Sie hier: zum Download Der Autor:Weitere Artikel online lesen


NewsSchnell und einfach die passende Trennsäule finden
© Text und Bild: Altmann Analytik ZEISS stellt neue Stereomikroskope vor
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen. © Text und Bild: Carl Zeiss Microscopy GmbH |