|
Neuer Alzheimer-Mechanismus lässt auf baldige Therapien hoffen
Neuer Alzheimer-Mechanismus lässt auf baldige Therapien hoffen
Die Alzheimer Demenz (AD) ist eine seit mehr als 100 Jahren beschriebene Erkrankung deren eigentliche Ursache bis heute noch immer nicht identifiziert ist, obwohl von aktuell mehr als 30 Mio. Erkrankten weltweit auszugehen ist. Neueste Hochrechnungen zeichnen ein dramatisches Szenario mit 106 – 360 Mio. Erkrankten im Jahre 2050. Es beginnt damit, dass man im Kaufhaus steht und plötzlich nicht mehr aus dem Laden findet oder einem der Weg nachhause nicht mehr einfallen will – zumindest ist dies oft der Zeitpunkt, an dem die meisten Betroffenen doch beschließen, zum Arzt zu gehen. Nach dem, was wir heute bereits wissen, beginnt die Krankheit allerdings schon sehr lange, bevor die ersten Symptome klinisch erkennbar werden.
Nervenzellverlust
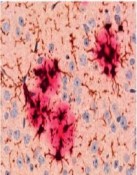
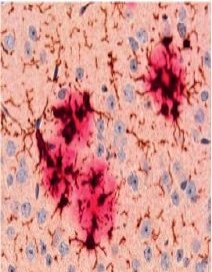
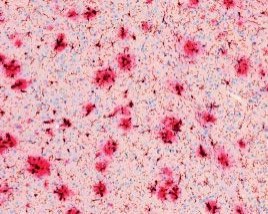
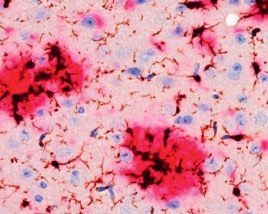
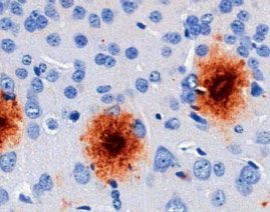
Im Laufe des Lebens reichern sich die Schnittprodukte des sog. Amyloid Precursor Proteins (APP, Amyloid-Vorläufer-Protein) im Gehirn an. Diese als „Aß“ bezeichneten Peptide gelten als jene, die eine Kaskade von verschiedenen pathologischen Ereignissen in Gang setzen und so letztlich zur AD führen. Dabei kommt es zunächst zur Zusammenlagerung der Peptide zu Aggregaten sehr unterschiedlicher Form und Größe. Es bilden sich lösliche Aggregate aus 2 bis etwa 12 Monomeren, die als oligomeres Aß bezeichnet werden. Daneben entstehen langkettige, unlösliche Strukturen, sog. Fibrillen, die sich zu einem der histologischen Hauptcharakteristika der AD zusammenlagern, den Aß-Plaques (Abb. 1). Die Plaques wie auch die neurofibrillären Tangles konnten schon 1905 von Alois Alzheimer durch Versilberung morphologisch sichtbar gemacht werden.
Genetische Analysen von erblichen Formen der AD (FAD) zeigten auch, dass diese Mutationen im APP-Gen oder 2 Prozessierungsenzymen des APP (Presenilin 1 oder 2) besitzen. Daher galten die Plaques noch bis vor wenigen Jahren als das eigentliche toxische Korrelat des Nervenzellverlustes. Erst im Jahr 2005 wurde erkannt, dass insbesondere das Dodekamer (12mer) des Aß extrem toxisch auf Neurone wirkt und Nagern das Gedächtnis „nimmt“. In den darauffolgenden Jahren wurde endgültig bestätigt, dass es die löslichen, monomeren und oligomeren Aß-Spezies sind, die diese Toxizitätsprobleme verursachen. Die Plaques sind vielmehr als Reaktion (Kristallisationskeim) anzusehen, die das lösliche Aß in großen Aggregaten binden.
*Risikofaktor Alter*
Wie aber kommt es dazu, dass sich zu viel Aß im Gehirn sammelt? Bei Patienten, die an der erblichen Form der AD leiden, führt eine erhöhte Produktion des APP oder dessen veränderte Prozessierung zur vermehrten Bildung von Aß. Diese Patienten machen jedoch weniger als 1 % aller Betroffenen aus. Über 99 % der Patienten erkranken an dieser sporadischen Form von Alzheimer (sAD) und für diese Variante der Krankheit konnte bislang nicht geklärt werden, warum sich zu viel Aß im Gehirn ansammelt. Als Risikofaktoren gelten das Rauchen, übermäßiger Alkoholgenuss, hoher Blutdruck und ein hoher Cholesterinspiegel –
 also eine ungesunde Lebensweise mit Gefäßproblemen im Allgemeinen. Ein erhöhtes Risiko haben zudem Menschen, die die sog E4-Variante des ApoE-Gens tragen. Das von diesem Gen kodierte Protein ist für den Transport von Fetten im Blut mitverantwortlich und trägt zur Regulierung des Cholesterinspiegels bei. Der mit Abstand größte bestimmte Risikofaktor ist heute noch immer das Alter. Die Wahrscheinlichkeit, an einer AD zu erkranken, verdoppelt sich ab dem 65. Lebensjahr etwa alle 5 Jahre, sodass im Alter von 90 Jahren mehr als ein Drittel aller Menschen an Alzheimer leidet.
also eine ungesunde Lebensweise mit Gefäßproblemen im Allgemeinen. Ein erhöhtes Risiko haben zudem Menschen, die die sog E4-Variante des ApoE-Gens tragen. Das von diesem Gen kodierte Protein ist für den Transport von Fetten im Blut mitverantwortlich und trägt zur Regulierung des Cholesterinspiegels bei. Der mit Abstand größte bestimmte Risikofaktor ist heute noch immer das Alter. Die Wahrscheinlichkeit, an einer AD zu erkranken, verdoppelt sich ab dem 65. Lebensjahr etwa alle 5 Jahre, sodass im Alter von 90 Jahren mehr als ein Drittel aller Menschen an Alzheimer leidet.
Wege zu früher Diagnostik
Das fehlende Wissen um den Entstehungsmechanismus ist auch ein Grund für den Mangel an diagnostischen Instrumenten. Ärzte sind zwar in der Lage, die Krankheit mithilfe klinischer Erfahrung und mit Kognitionstests gut von anderen Demenzformen abzugrenzen, jedoch erst in einem Stadium, in dem eine Behandlung eigentlich schon zu spät kommt. Denn wenn auch in den letzten Jahren klar wurde, dass das Gehirn eine größere Fähigkeit zur Regeneration besitzt als ursprünglich angenommen, ist der Schaden in diesem Stadium der AD bereits viel zu groß, um die zerstörten Nervenzellen vollständig ersetzen zu können. Vor allem mithilfe von Substanzen, die an das Aß binden und mittels Positronen-Emissions-Tomografie (PET) nachgewiesen werden können und der Bestimmung von Aß 40 und 42 sowie von phosphoryliertem Tau-Protein im Nervenwasser der Patienten wird derzeit versucht, eine frühere Diagnostik zu erreichen. Diese Verfahren sind viel versprechend, aber leider bisher noch nicht gut genug, um eine effektive Frühdiagnostik zu ermöglichen. Ziel wäre es, die Krankheit in einem Stadium zu diagnostizieren, in dem noch gute Behandlungserfolge möglich wären, wenn zukünftig eine effektive Behandlung verfügbar sein wird.
Transportproteine im Visier
Um all diese Anforderungen erfüllen zu können, braucht es in erster Linie einen pathogenetischen Mechanismus, der die sporadische Form der AD erklärt. Spätestens als im Dezember 2010 eine amerikanische Studie der University of Washington veröffentlicht wurde [1], in der gezeigt werden konnte, dass die Produktion von Aß in ADPatienten nicht höher ist als in Gesunden, kam die
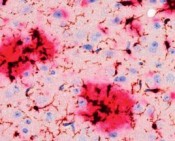 Aß-Produktionshypothese stark ins Schwanken. In der gleichen Arbeit wurde allerdings auch gezeigt, dass offensichtlich der Abtransport des Aß aus dem Gehirn bei erkrankten Personen um 30 % reduziert ist. Nun wird schon seit einigen Jahren spekuliert, dass ein gestörter Abtransport von Aß zur AD führen könnte. Verantwortlich dafür wurde vor allem das Protein LRP1 gemacht, das auch im Fettstoffwechsel eine Rolle spielt. Jedoch blieben die Erfolge aus, dies als therapeutisches Ziel zu nutzen. Vor etwa 10 Jahren wurde jedoch mit dem ABCB1 Protein eine weitere Proteinsuperfamilie zu den möglichen Aß-Exportproteinen hinzugefügt. Das Forschungsinteresse war bisher nur sehr gering, trotz weiterer Hinweise darauf, dass das ABCB1 eine wesentliche Rolle bei der Erkrankung spielen könnte. ABCB1 gehört zur Superfamilie der ATP-binding-cassette-Transporter (ABC-Transporter). Diese Transportproteine haben
Aß-Produktionshypothese stark ins Schwanken. In der gleichen Arbeit wurde allerdings auch gezeigt, dass offensichtlich der Abtransport des Aß aus dem Gehirn bei erkrankten Personen um 30 % reduziert ist. Nun wird schon seit einigen Jahren spekuliert, dass ein gestörter Abtransport von Aß zur AD führen könnte. Verantwortlich dafür wurde vor allem das Protein LRP1 gemacht, das auch im Fettstoffwechsel eine Rolle spielt. Jedoch blieben die Erfolge aus, dies als therapeutisches Ziel zu nutzen. Vor etwa 10 Jahren wurde jedoch mit dem ABCB1 Protein eine weitere Proteinsuperfamilie zu den möglichen Aß-Exportproteinen hinzugefügt. Das Forschungsinteresse war bisher nur sehr gering, trotz weiterer Hinweise darauf, dass das ABCB1 eine wesentliche Rolle bei der Erkrankung spielen könnte. ABCB1 gehört zur Superfamilie der ATP-binding-cassette-Transporter (ABC-Transporter). Diese Transportproteine haben
sich in der Evolution in allen Eukaryoten durchgesetzt, weil sie den Organismus vor einer enormen Vielfalt giftiger Umweltstoffe und Stoffwechselprodukte schützen, indem sie durch Aufwendung von Energie, ATP, ihre Substrate aus den Zellen bzw. aus dem Organismus hinaustransportieren – und das auch gegen große Konzentrationsgradienten. Nachdem wir bei der Untersuchung von Hirnschnitten gesunder Patienten eine inverse Korrelation von Aß-Ablagerungen und ABCB1-Protein in den Hirnkapillaren gefunden hatten [2], wollten wir das Phänomen genauer biochemisch und molekularbiologisch untersuchen und begannen mit der Züchtung von transgenen Mauslinien, die die Aß-Pathologie der AD entwickeln, denen jedoch zusätzlich verschiedene ABC-Transporter fehlen (ABC Transporter knock outs). Das Ergebnis ist eine substanzielle Anzahl weltweit einmaliger Mausstämme, deren Analyse den enormen Einfluss dieser Proteine auf die Akkumulation von Aß im Gehirn zeigte [3]. So stellte sich nicht nur heraus, dass sich in ABCB1- knockout-Tieren im Laufe der ersten 25 Lebenswochen die 3,5-fache Menge an Aß im Vergleich zu normalen Kontrolltieren ansammelt, sondern vielmehr, dass der Transporter ABCC1 eine offensichtlich wesentlich größere Rolle in diesem Exportsystem spielt. Bei ABCC1-defizienten Tieren haben wir im gleichen Alter eine 12- bis 14-fache Menge Aß im Gehirn nachweisen können. Diesen Effekt konnten wir nicht nur in einem weiteren Tiermodell finden, bei dem sich die Ablagerungen fast ausschließlich in den Blutgefäßen manifestieren, sondern mithilfe eines mathematischen Modells gezielt beweisen und vorhersagen. Anhand dieser Berechnungen konnten wir sehen, dass der ca. 4-fache Unterschied zwischen den ABCB1- und ABCC1-knockout-Tieren einer Verringerung der Transportrate um nur 11 % entspricht. Das bedeutet, dass eine minimale Verringerung der Exportkapazität über einen langen Zeitraum verheerende Folgen haben kann.
Transporter mit Potenzial
In diesem Zusammenhang ist es enorm wichtig, sich in Erinnerung zu rufen, welche physiologische Aufgabe (gerade diese beiden) ABC-Transporter haben. Sie sind hauptsächlich dafür verantwortlich zu machen, dass die meisten der Medikamente, die wir einnehmen, nicht ins Gehirn gelangen. Dieser Schutz hat jedoch gleichzeitig zur Folge, dass Transportkapazität zum Beispiel für das Aß nicht mehr vollständig zur Verfügung steht. Wenn man gleichzeitig noch eine sich verschlechternde Energieversorgung durch verringerte Durchblutung und alternde Mitochondrien hinzurechnet, entsteht ein Bild, das die Pathogenese der sporadischen AD mit erklären könnte. Ein Mechanismus mit derart starkem Einfluss bietet aber gleichzeitig ein hohes therapeutisches und diagnostisches Potenzial. Ist man in der Lage, die Aktivität von ABC-Transportern an der Blut-Hirn-Schranke gezielt zu erhöhen, hätte man eine viel versprechende Therapieoption. Nun ist es allerdings so, dass ABC-Transporter aufgrund ihrer evolutionären Natur bisher eher als Hindernis angesehen wurden, Medikamente
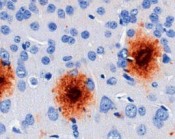 an ihren Bestimmungsort zu bringen. Gerade in der Krebstherapie sind diese Proteine ein Dorn im Auge, da sie die Resistenz der Krebszellen gegen die Chemotherapeutika verursachen. Aus diesem Grund wurden fast ausschließlich Inhibitoren für die ABC-Transporter entwickelt und den Aktivatoren wenig Aufmerksamkeit gewidmet. Dennoch ist es uns gelungen ein Medikament (Thiethylperazin) zu finden, das die Aktivität von ABCC1 um etwa 70 % erhöht. Mäuse, die mit Thiethylperazin prophylaktisch (vor Beginn) und therapeutisch (nach Beginn der AD) behandelt wurden, hatten nach jeweils 25 Tagen weniger als die Hälfte Aß im Gehirn als unbehandelte Tiere. Aufgrund dieser Ergebnisse werden wir nun untersuchen, ob eine Aktivierung von ABCC1 grundsätzlich als Therapie bei AD-Patienten einsetzbar ist. Darüber hinaus wollen wir diesen Mechanismus für die Diagnostik nutzbar machen, denn bei Nachweis einer verminderten Transportkapazität von ABCC1 bei spezifischen Patienten kann man frühzeitig Gegenmaßnahmen ergreifen, die so die Erkrankung bis in ein höheres Alter hinauszögern könnten.
an ihren Bestimmungsort zu bringen. Gerade in der Krebstherapie sind diese Proteine ein Dorn im Auge, da sie die Resistenz der Krebszellen gegen die Chemotherapeutika verursachen. Aus diesem Grund wurden fast ausschließlich Inhibitoren für die ABC-Transporter entwickelt und den Aktivatoren wenig Aufmerksamkeit gewidmet. Dennoch ist es uns gelungen ein Medikament (Thiethylperazin) zu finden, das die Aktivität von ABCC1 um etwa 70 % erhöht. Mäuse, die mit Thiethylperazin prophylaktisch (vor Beginn) und therapeutisch (nach Beginn der AD) behandelt wurden, hatten nach jeweils 25 Tagen weniger als die Hälfte Aß im Gehirn als unbehandelte Tiere. Aufgrund dieser Ergebnisse werden wir nun untersuchen, ob eine Aktivierung von ABCC1 grundsätzlich als Therapie bei AD-Patienten einsetzbar ist. Darüber hinaus wollen wir diesen Mechanismus für die Diagnostik nutzbar machen, denn bei Nachweis einer verminderten Transportkapazität von ABCC1 bei spezifischen Patienten kann man frühzeitig Gegenmaßnahmen ergreifen, die so die Erkrankung bis in ein höheres Alter hinauszögern könnten.
Literatur
[1] Mawuenyega, K. G. et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science 330, 1774 (2010).
[2] Vogelgesang, S. et al. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer's disease. Curr Alzheimer Res 1, 121 – 125 (2004).
[3] Krohn, M. et al. Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. J Clin Invest 121, 3924 – 3931(2011).
Foto: © Prof. Dr. Dr. Jens Pahnke
|
L&M 6 / 2011
Diese Artikel wurden veröffentlicht in Ausgabe L&M 6 / 2011.
Das komplette Heft zum kostenlosen Download finden Sie hier:
zum Download
Die Autoren:
Weitere Artikel online lesen
News
Mit dem HPLC-Säulenkonfigurator unter www.analytics-shop.com können Sie stets die passende Säule für jedes Trennproblem finden. Dank innovativer Filtermöglichkeiten können Sie in Sekundenschnelle nach gewünschtem Durchmesser, Länge, Porengröße, Säulenbezeichnung u.v.m. selektieren. So erhalten Sie aus über 70.000 verschiedenen HPLC-Säulen das passende Ergebnis für Ihre Anwendung und können zwischen allen gängigen Herstellern wie Agilent, Waters, ThermoScientific, Merck, Sigma-Aldrich, Chiral, Macherey-Nagel u.v.a. wählen. Ergänzend stehen Ihnen die HPLC-Experten von Altmann Analytik beratend zur Seite – testen Sie jetzt den kostenlosen HPLC-Säulenkonfigurator!
© Text und Bild: Altmann Analytik
Aufnahme, Dokumentation und Teilen von Ergebnissen mit ZEISS Stemi 305 und ZEISS Stemi 508
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen.
© Text und Bild: Carl Zeiss Microscopy GmbH
|