


|
Molekulare Epidemiologie bakterieller Krankheitserreger
Molekulare Epidemiologie bakterieller KrankheitserregerAbgrenzung zufälliger Häufungen von Übertragungen und AusbruchsgeschehenImmer häufiger hört man in den Medien von Infektionsausbrüchen, im Mittelpunkt stehen hier Ausbrüche mit multiresistenten bakteriellen Erregern (MRE). Doch woher kommen diese Erreger und was sind mögliche Ursachen für deren Anstieg? Wie kann man bei gehäuftem MRE-Auftreten zufällige Häufungen von Ausbrüchen differenzieren? Der folgende Artikel erklärt Methoden, mit denen Häufungsgeschehen analysiert und die molekulare Epidemiologie aufgeklärt werden können. Entstehung und Weiterverbreitung von multiresistenten Erregern 
Die Entstehung und Weiterverbreitung von MRE wird von verschiedenen Faktoren begünstigt. Wichtigster Grund für die Entstehung von antibiotikaresistenten Erregervarianten ist der Selektionsdruck durch Antibiotika, d. h., bei jeder Gabe eines Antibiotikums haben antibiotikaresistente Erregervarianten, die z. B. durch Mutationen oder Übertragung von Resistenzgenen entstehen, einen Wachstumsvorteil. Verschärft wird die Situation durch einen Anstieg der Antibiotikaverschreibungen in den letzten Jahren (www.p-e-g.org/
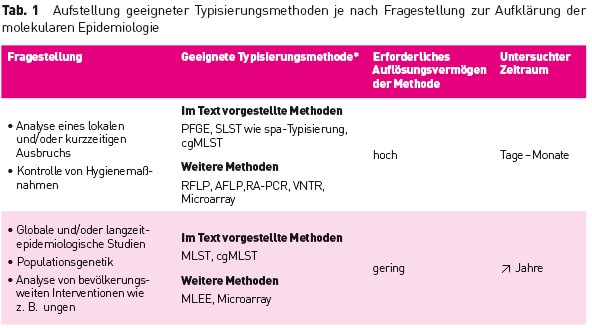
Tab.1 Aufstellung geeigneter Typisierungsmethoden je nach Fragestellung zur Aufklarung der molekularen Epidemiologie
Werkzeug zur Aufklärung der molekularen Epidemiologie – die Typisierung
Die Erregertypisierung ist das wichtigste Werkzeug, um herauszufinden, ob mehrere ähnliche Erreger einer epidemiologisch identifizierten Häufung gleich sind und damit eine Übertragung stattgefunden hat. Hierzu wurden verschiedene phänotypische und genotypische Verfahren entwickelt [1], die sich durch unterschiedliche Charakteristika auszeichnen und meist nur für spezielle Fragestellungen (z. B. Ausbruchsuntersuchungen, Evolutionsstudien) besser oder weniger gut geeignet sind (Tab. 1). Grundannahme aller Typisierungen, bei der je nach Verfahren verschiedene Charakteristika des Erregers untersucht werden, ist, dass identische Erreger ein gleiches Ergebnis liefern oder zumindest eine hohe Ähnlichkeit aufweisen. In diesen Fällen ist von einer Übertragung auszugehen. Dabei sollte jedes Typisierungsverfahren reproduzierbare Ergebnisse liefern, um auch eine Vergleichbarkeit über Laborgrenzen hinweg zu ermöglichen. Darüber hinaus ist – zumindest für Ausbruchsanalysen – ein möglichst hohes Auflösungsvermögen erstrebenswert, um auch bei nah verwandten Erregern eine sichere Abgrenzung zu erreichen. Schließlich sollten die Ergebnisse zur epidemiologischen Situation konkordant sein [1].
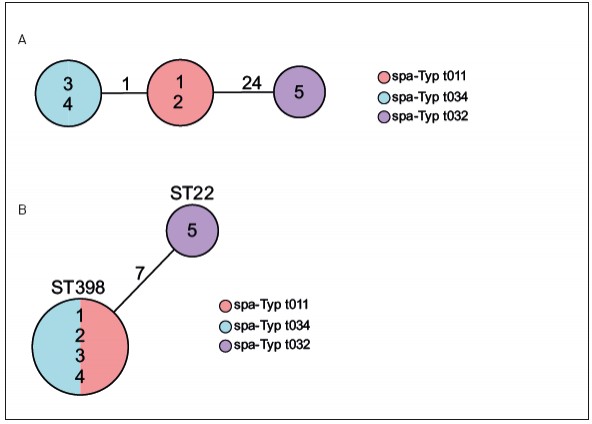
Abb.1 Typisierung von funf MRSA-Isolaten aus einem lokalen Haufungsgeschehen. Die Isolatbezeichnungen sind jeweils in den Kreisen der Diagramme angegeben. (A) zeigt die Verwandtschaftsverhaltnisse der funf MRSA-Isolate nach spa-Typisierung. Hierbei entspricht jeder Kreis einem identischen Genotyp (spa-Typ) und ist entsprechend eingefarbt. Die Zahlen auf den Verbindungslinien zwischen zwei Kreisen geben die Anzahl der evolutiven Schritte an, die zwischen zwei spa-Typen liegen, und sind ein Mas des Verwandtschaftsgrades. Je niedriger der Wert, desto hoher der Verwandtschaftsgrad. (B) zeigt die Verwandtschaftsverhaltnisse der funf MRSA-Isolate nach MLST in einem Minimum Spanning Tree. Jeder Kreis ist ein Genotyp (ST), der auf einem Allelprofil von sieben Genen beruht, und entsprechend benannt. Die Zahl auf der Verbindungslinie zweier Kreise gibt die Zahl unterschiedlicher Allele von den insgesamt sieben untersuchten Genen an. Zusatzlich sind die Kreise nach ihren spa-Typen eingefarbt.
Beispiel MRSA – Aufklärung der molekularen Epidemiologie Wahrscheinlich der bekannteste MRE ist der methicillinresistente Staphylococcus aureus (MRSA). Bei gehäuftem Auftreten besteht die Herausforderung darin, die molekulare Epidemiologie dieser Erreger zu bestimmen, um anhand dessen zwischen einer zufälligen Häufung und einem Ausbruch, der durch Übertragung von einem zum anderen Menschen verursacht wurde, zu unterscheiden. In diesem Beispiel gibt es fünf Patienten, die in einem Krankenhaus einen MRSA-Nachweis hatten, wodurch der Verdacht von Übertragungen nahelag. Diese MRSA wurden spa-typisiert (Abb. 1A). Hierbei zeigte sich, dass es drei verschiedene spa-Typen gab (t011, t034, t032), womit eine Übertragung von Patient zu Patient in drei der fünf Fälle sicher ausgeschlossen werden konnte. Wenn man diese Isolate mittels MLST untersuchen würde, ließen sich vier der fünf Isolate nicht voneinander unterscheiden; lediglich Isolat 5 hatte einen anderen Sequenztyp (Abb. 1B). An diesem Beispiel zeigt sich sehr deutlich das unterschiedliche Auflösungsvermögen verschiedener Methoden. Was macht man aber bei den Patienten, die einen identischen spa-Typ haben? Hier kann ja eine Übertragung nicht ausgeschlossen werden, sodass hier entweder Maßnahmen für einen Ausbruch wie z. B. verschärfte Hygienemaßnahmen etabliert werden müssen oder intensive epidemiologische Recherchen die Möglichkeit einer Übertragung noch einmal überprüfen und dann ggf. doch ausschließen können. Bis vor Kurzem war diese Situation häufig der Endpunkt. Aufgrund der rasanten technischen Weiterentwicklung im Bereich der Next-Generation- Sequenzierungstechnologie ist es seit einigen Jahren möglich, das Auflösungsvermögen dramatisch zu verstärken, da nicht nur einzelne Gene, sondern ganze Genome sequenziert und deren Information auch zur Aufklärung der molekularen Epidemiologie eingesetzt werden können. Eine Analysemöglichkeit, die als „core genome MLST“ (cgMLST) bezeichnet wird [9], funktioniert in Analogie zur klassischen MLST: auch hier wird jedem Gen ein Allel zugeordnet und die Allele aller analysierten Gene bilden dann ein Allelprofil, welches das Typisierungsergebnis darstellt. Da jetzt aber Daten von weit über 1.000 Genen vorliegen, kann man ein Auflösungsvermögen erreichen, das die Erregerausbreitung mit maximaler Genauigkeit widerspiegelt. Abbildung 2 zeigt, dass von den fünf Patientenisolaten zwei (1 und 2) identisch waren und damit eine Übertragung als gesichert gewertet wurde. In den übrigen Fällen konnte eine Übertragung aufgrund der Unterschiede ausgeschlossen werden (Abb. 2). Aufgrund des hohen Auflösungsvermögens ist es zukünftig denkbar, diese Typisierung z B. in einem Krankenhaus flächendeckend prospektiv für alle MRE einzusetzen, um frühzeitig eine Erregerausbreitung zu detektieren. Nur wenn sehr ähnliche oder identische Erreger gefunden werden, sind ggf. weitere epidemiologische Recherchen notwendig. Hygienemaßnahmen können so gezielter eingesetzt werden und die knappen Ressourcen schonen.
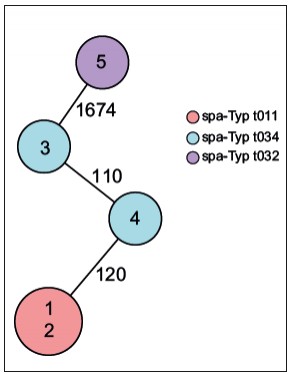
Abb.2 Darstellung der Verwandtschaftsverhaltnisse der funf MRSA-Isolate mittels Core-genome- MLST-Analyse [10]. In diesem Minimum Spanning Tree ist jeder Kreis ein Genotyp basierend auf dem Allelprofil von 1.861 Genen und entsprechend mit einer Isolatnummer gekennzeichnet. Die Zahl auf der Verbindungslinie zweier Kreise gibt die Anzahl unterschiedlicher Allele von den insgesamt 1.861 untersuchten Genen an. Die Kreise sind zusatzlich je nach spa-Typ eingefarbt und die Grose der Kreise ist proportional zur Anzahl der Isolate mit identischem Genotyp. In diesem Beispiel sind die Isolate 1 und 2 identisch, was fur eine Ubertragung spricht.
-> alexander.mellmann@ukmuenster.de
Bild: © istockphoto.com| perkmeup |
L&M 2 / 2016
Das komplette Heft zum kostenlosen Download finden Sie hier: zum Download Die Autoren:Weitere Artikel online lesen


NewsSchnell und einfach die passende Trennsäule finden
© Text und Bild: Altmann Analytik ZEISS stellt neue Stereomikroskope vor
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen. © Text und Bild: Carl Zeiss Microscopy GmbH |





