

|
Medizin
 >
Die Nutzung von Protein-Microarrays zur personalisierten Proteomanalyse
>
Die Nutzung von Protein-Microarrays zur personalisierten Proteomanalyse
Die Nutzung von Protein-Microarrays zur personalisierten ProteomanalyseEinmal durchchecken, bitte!Nach der Entschlüsselung der menschlichen Erbsubstanz und der Möglichkeit, die in der Basensequenz enthaltenen individuellen Unterschiede für klinische Routinediagnostik zu nutzen, ist mittlerweile die Analyse einer weiteren, wesentlich komplexeren und therapie-näheren Molekülklasse – der Proteine – soweit voran geschritten, eine personenbezogene, sprich „personalisierte“ Diagnosestellung zu unterstützen. 
Zurzeit erhalten wir einen Einblick, wie die Möglichkeit zu einer vollständigen und gleichzeitig für eine klinische Anwendung ausreichend genauen Analyse der menschlichen Erbsubstanz – des Genoms – die Diagnostik umwälzen dürfte. Die Kapazität, in jedem Individuum alle jeweiligen Varianten der Genomsequenz auszulesen und medizinisch relevante Schlussfolgerungen für diese Person zu ziehen, wird die Stratifizierung von Patienten – eine Einteilung in Untergruppen, die mit verschiedenen Therapieformen behandelt werden sollten – enorm vorantreiben. Gleichzeitig bietet sich eine Chance, bei Krankheiten mit sehr komplexem molekulargenetischen Hintergrund wie etwa Krebs die Kombination der Veränderungen zu finden, die für das individuelle Krankheitsbild verantwortlich sind. Noch genauere Information sollte gerade auch für nicht genetisch bedingte Krankheiten eine Analyse aller Proteine eines Gewebes – des Proteoms – bieten, da Proteine im Gegensatz zu den meisten Nukleinsäuren unmittelbar an der molekularen Umsetzung der im Genom kodierten Information beteiligt sind. Das ist auch ein Grund, warum die meisten zurzeit genutzten Wirkstoffe in Medikamenten Proteine beeinflussen. Gerade auch für das sich entwickelnde Feld der Companion Diagnostics, eines einem Wirkstoff zugeordneten diagnostischen Verfahrens, das die Information liefert, ob dieser Wirkstoff für den individuellen Patienten sicher ist und einen positiven Effekt auf den Krankheitsverlauf haben wird, ist deshalb eine genaue Information über das beeinflusste Protein und sein Umfeld grundlegend. Im letzten Jahrzehnt hat die Proteomanalyse einen enormen Sprung nach vorn gemacht. Ganz speziell die vielen Analysewege mittels Massenspektrometrie haben das Feld revolutioniert. Erst vor Kurzem wurden erste, grobe Zusammenstellungen des menschlichen Proteoms veröffentlicht [1–3]. Wie jedoch schon bei der Sequenzierung des Genoms beginnt erst jetzt die Nutzung dieser Information, um sie in klinische Anwendungen zu übersetzen. Dabei sind die Anforderungen um ein Vielfaches größer als bei der DNA-Sequenzierung. Einmal besitzt das Proteom eine enorme Komplexität. Die etwa 22.000 Gene des Menschen werden in geschätzt eine bis mehrere Millionen Proteinformen umgeschrieben; so gibt es im Menschen beispielsweise 566 bekannte Proteasen, die andere Proteine schneiden und damit in Struktur und Funktion verändern können. Dazu kommt eine noch unbekannte Zahl krankheitsbedingter Variationen. Zusätzlich sind Proteine – im Gegensatz zu Nukleinsäuren – von ihrer Struktur und Biochemie sehr unterschiedlich und variieren stark in Konzentration und Lokalisierung.
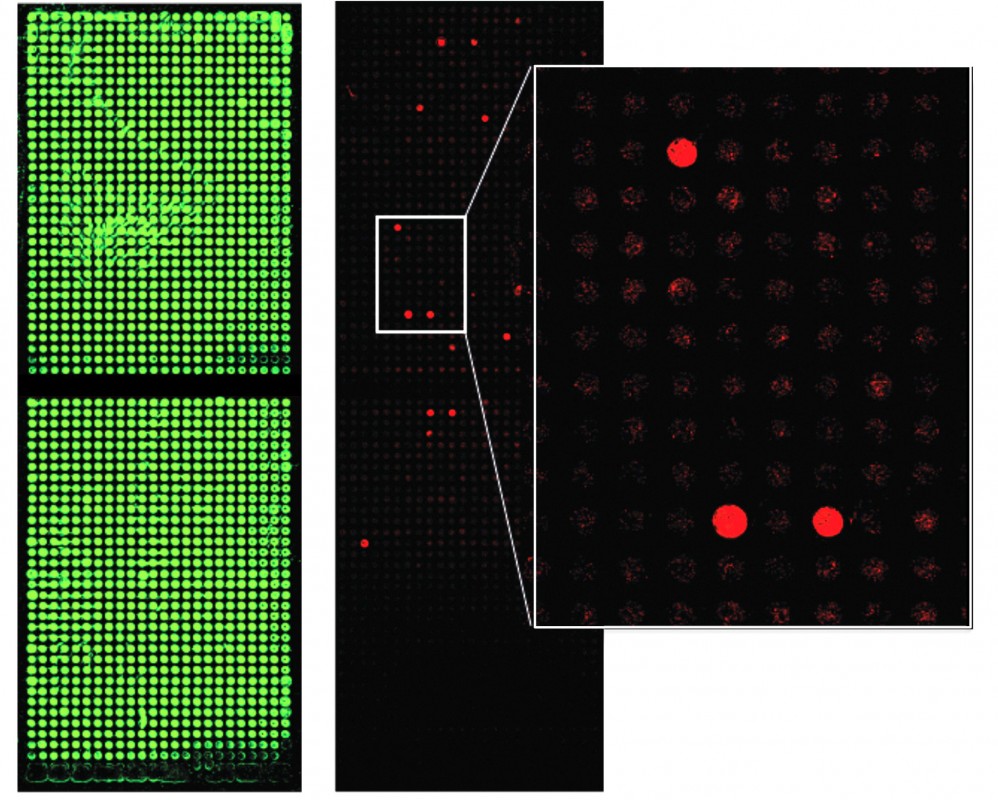
Abb. Protein-Microarray mit mehr als 1600 verschiedenen Proteinen, die durch zellfreie In-situ-Synthese produziert wurden. Links sind alle Proteine nach einer Färbung mit einem grünen Fluoreszenzfarbstoff zu erkennen. Rechts ist das Ergebnis einer Inkubation mit Antikörpern zu sehen, die mit rotem Farbstoff markiert waren (Lueong und Hoheisel, unpublizierte Ergebnisse).
Tanz der Moleküle Von ganz besonderer Wichtigkeit ist jedoch die Tatsache, dass die meisten Proteine mit anderen Proteinen oder Liganden interagieren, um ihre Funktion auszuüben. Im Gegensatz zu der ebenfalls wichtigen Interaktion zwischen Nukleinsäuren, die nach relativ einfachen und leicht zu definierenden Regeln verläuft, ist bei Proteinen quasi jede Interaktion unterschiedlich. Dabei sind Stärke und Dauer der Bindung von wesentlicher Bedeutung. Deshalb sind qualitative Verfahren wie das Yeast-Two-Hybrid-System hervorragend geeignet, Interaktionen zu identifizieren, erlauben jedoch keine Analyse im diagnostischen Bereich. Es werden Methoden benötigt, die diese Interaktionen zumindest semi-quantitativ und in großer Zahl messbar machen. Die Intensität einer Interaktion wird durch die Affinität zwischen zwei Molekülen definiert, die wiederum durch Faktoren wie Struktur, Ladung oder Konzentration beeinflusst wird. Affinitätsgestützte Proteindiagnostik ist in der Medizin bereits lange fest etabliert. Verfahren wie Immunhistochemie oder ELISA sind aus der klinischen Diagnostik nicht mehr wegzudenken. Gerade um komplexe biologische Vorgänge beschreiben und analysieren zu können, sind jedoch andere Verfahren notwendig. Protein-Microarrays sind eine solche Plattform [4]. Wie die Massenspektrometrie ist sie methodisch mittlerweile zumindest in Teilbereichen von einer für klinische Diagnostik ausreichenden Qualität. Dabei liefert sie Informationen, die vielfach komplementär zu den Ergebnissen der Massenspektrometrie sind. Die Nachweisgrenze ist grundsätzlich besser als das, was ELISA oder Massenspektrometrie zurzeit leisten können, bis zum Nachweis auf Einzelmolekülebene. Gleichzeitig können prinzipiell beliebig viele Messungen auf einmal durchgeführt und ausgelesen werden. Protein-Microarrays lassen sich in drei Grundformate unterteilen. Einmal sind viele, verschiedene Proteine auf dem Microarray individuell präsentiert. Eine Sonderform davon sind Antikörper-Microarrays, quasi eine Muliplexversion des ELISA, die jedoch eine Reihe an Vorteilen mit sich bringt. Bei der dritten Variante werden Proteinextrakte aus Patientenmaterial in ihrer Gesamtheit auf einen Träger aufgebracht und miteinander vergleichend analysiert. Auf die richtige Bindung kommt es an Microarrays vieler verschiedener Proteine können durch das Aufdrucken vorgefertigter Moleküle oder deren In-situ-Synthese produziert werden. Zum Nachweis von Immunantworten des Patienten auf Infektionen, Allergien oder anderer Erkrankungen sind beide Formate bereits präklinisch im Einsatz. Eine Analyse des Proteoms eines individuellen Patienten lässt sich sinnvoll nur durch In-situ-Synthese der Proteine erreichen. In unserem Labor wurde dazu eine Methode entwickelt, die aus der Gesamt-RNA des betroffenen Gewebes auf dem Microarray DNA-Kopien der individuellen RNA-Moleküle herstellt, die alle Mutationen oder Reifungsvarianten der RNA beinhalten. Von dieser DNA werden durch zellfreie Transkription und Translation die Proteinvarianten produziert, die im Patienten vorliegen. Deren Auswirkungen können somit individuell getestet werden. Methodisch am weitesten fortgeschritten ist die Nutzung von Antikörper-Microarrays. Grundsätzlich können alle Proteine des Körpers daraufhin untersucht werden, in welchen Mengen, Strukturen und mit welchen Modifikationen sie vorliegen. Voraussetzung ist das Vorhandensein guter Antikörper gegen die jeweilige Zielstruktur (Epitop). Obwohl zurzeit in der Datenbank Antibodypedia mehr als 1,4 Mio. Antikörper gegenüber 19.000 Genprodukten aufgelistet sind, fehlen für viele Anwendungen noch passende Antikörper oder andere, entsprechende Bindemoleküle. Speziell für Strukturanalysen sind beispielsweise nur wenige Bindemoleküle vorhanden. Dies liegt u.a. auch daran, dass IgG-Antikörper hierfür wahrscheinlich nur unzureichend geeignet sind, da sie meistens lineare Epitope erkennen. Andere Bindertypen wie etwa Nanobodies – Antikörperderivate aus Kamelen oder Haien – zeigen dagegen eine sehr hohe strukturspezifische Erkennung. Dies kann übrigens für andere Testverfahren wie Immunhistochemie wiederum von Nachteil sein, da die Proteine dort vielfach denaturiert vorliegen. Für eine vollständige Analyse des Proteoms ist folglich eine Vielzahl neuer und/oder besserer Binder gegen bisher nicht abgedeckte Epitope notwendig. Weltweit ist dies zurzeit ein Schwerpunkt vieler akademischer und industrieller Anstrengungen [5]. Microarrays aus vollständigen Proteinextrakten stellen eine vereinfachte Version von Gewebeschnitten dar. Durch Inkubation mit Antikörpern kann beispielsweise festgestellt werden, ob ein bestimmtes Protein im Gewebe vorliegt. Die vereinfachte Messung und der erhöhte Durchsatz werden jedoch durch eine Reduzierung des Informationsgehalts erkauft. Aus Gewebeschnitten lassen sich neben der reinen Mengenangabe auch Informationen über die Lokalisierung und Verteilung des Proteins ablesen. Insgesamt befinden sich Protein-Microarrays zurzeit aufgrund der steten Entwicklung in einer Übergangsphase – zumindest für einige wenige Anwendungen – von der rein forschungsorientierten Nutzung zur Anwendung an Patientenmaterial hin zur klinischen Diagnose. Der Aspekt der Zertifizierung ist dabei eine schwierige Frage. Zurzeit wird häufig davon ausgegangen, dass ein klinischer Test möglichst einfach aufgebaut sein sollte, etwa wie ein Schwangerschaftstest. Aufgrund der Komplexität vieler Krankheitsbilder kann es aber gut möglich sein, dass nur eine breit ausgelegte Proteomanalyse zu einer ausreichend genauen Diagnose führen kann, da nur hierdurch molekulare Kompensationsvorgänge erfasst werden und in die Diagnose einfließen können. Und wie für alle Verfahren der Proteomanalyse – auch der Massenspektrometrie – ist das Vorliegen einer ausreichenden Zahl gut definierter Antikörper oder äquivalenter Moleküle eine Grundvoraussetzung.
Literatur Bild: © istockphoto.com| xavigm |
L&M 10 / 2014
Das komplette Heft zum kostenlosen Download finden Sie hier: zum Download Der Autor:Weitere Artikel online lesen



NewsSchnell und einfach die passende Trennsäule finden
© Text und Bild: Altmann Analytik ZEISS stellt neue Stereomikroskope vor
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen. © Text und Bild: Carl Zeiss Microscopy GmbH |





