


|
Vom Genom über das Proteom
Vom Genom über das ProteomZum Verständnis des Lebens am Beispiel des Infektionserregers Staphylococcus aureusMultiresistente Stämme von Staphylococcus aureus und anderen Bakterien stellen eine zunehmende Bedrohung der Menschheit dar; Ärzte, Wissenschaftler und Politiker sind sich einig: Es werden dringend neue Antibiotika, Vakzinierungsansätze sowie alternative Antiinfektionsstrategien benötigt, wenn wir nicht in die Zeit vor der Einführung der Antibiotika zurückfallen wollen. Mithilfe der neuartigen Möglichkeiten der modernen Genomforschung möchten wir zu einer umfassenden Kenntnis der Physiologie und Pathophysiologie der Staphylokokken gelangen, um sie nicht nur besser verstehen, sondern auch bekämpfen zu können. Wir forschen gemeinsam mit Kolleginnen und Kollegen aus Greifswald, Münster, Tübingen und Würzburg im Sonderforschungsbereich/Transregio 34, der von der Deutschen Forschungsgemeinschaft gefördert wird. Erste Ergebnisse dieses hoch ambitionierten, wichtigen Vorhabens werden hier vorgestellt. Multiresistente Bakterien – eine Bedrohung der Menschheit 
Multiresistente Stämme von Staphylococcus aureus stellen eine zunehmende Bedrohung der Menschheit dar (Abb. 1). Diese gefährlichen Bakterien sind nicht nur für ein Drittel der gefürchteten Krankenhausinfektionen verantwortlich, sie können auch sonst schwere Erkrankungen wie Endokarditis oder Sepsis auslösen. Besonders problematisch: Wegen der zunehmenden und im hohen Maße besorgniserregenden Resistenz gegenüber verschiedenen Antibiotika zeigen oft nur noch wenige Medikamente die erhoffte Wirkung. Obwohl die Fachleute vor dieser Entwicklung seit geraumer Zeit nachhaltig warnen, ist immer noch keine Abhilfe in Sicht. Inzwischen kennen wir Bakterien, die durch keines der vorhandenen Antibiotika zu therapieren sind, eine Situation, die uns in erschreckender Weise an die Zeit vor der Einführung der Antibiotika erinnert. Nicht nur Fachleute, in jüngster Zeit endlich auch Politiker sind sich darin einig: Es muss dringend etwas passieren, wenn eine Katastrophe für die Menschheit verhindert werden soll. Dabei stehen neben neuen Antibiotika auch Vakzinierungsansätze sowie alternative Antiinfektionsstrategien wie eine generelle Stärkung des Immunsystems im Fokus des Interesses [1]. Eine Vision hat uns in Greifswald im Zeitalter der Genomics und Postgenomics nicht mehr losgelassen: Mithilfe der neuartigen Möglichkeiten der Genomforschung wollen wir zu einem völlig neuen und umfassenden Verständnis der Lebensprozesse pathogener Bakterien gelangen, nicht nur im Schüttelkolben im Labor, sondern auch im Infektionsprozess im Krankenhaus. Wenn wir das bakterielle Leben besser verstehen, werden wir auch lernen, die Infektionserreger wirksamer zu bekämpfen. Das war der Ausgangspunkt für den in Greifswald vor einigen Jahren gemeinsam mit Infektionsbiologen und Medizinern in Würzburg (Hacker), Tübingen (Götz und Peschel) und später auch in Münster (Peters) auf den Weg gebrachten Sonderforschungsbereich/Transregio 34 der DFG zum Thema „Pathophysiology of staphylococci in the post-genomic era“ (2006–2018). Dieser verfolgt das Ziel, durch die gezielte Anwendung des neuen Methodenarsenals der funktionellen Genomforschung, in erster Linie der Proteomics, das Leben der Pathogene, ihren Stoffwechsel, ihre Anpassung an die wachstumsbegrenzenden Faktoren, die sie im Wirt vorfinden, ihr Virulenzpotenzial, mit dem sie den Wirt versuchen zu bekämpfen, ihre Strategien, das humane Immunsystem zu umgehen und sich zu schützen, und viele andere Aspekte ihrer Pathophysiologie umfassender zu verstehen, um mit diesem neuen Wissen ausgestattet, neue Bekämpfungsstrategien abzuleiten. Das ist natürlich ein sehr ambitioniertes Vorhaben, das einen langen Atem verlangt! Die genomische Revolution – oder Leben in seiner Gesamtheit verstehen
Wir sind gegenwärtig Zeugen einer Entwicklung, die sehr treffend mit dem Begriff „genomische Revolution“ bezeichnet wird und die zu einem Paradigmenwechsel in den Lebenswissenschaften geführt hat. Ausgangspunkt dieser neuen Entwicklung war die Publikation der ersten vollständigen Genomsequenz des Bakteriums Haemophilus influenzae im Jahre 1995. Nur sechs Jahre später folgte die humane Genomsequenz, der Öffentlichkeit vorgestellt im Weißen Haus zu Washington zum Thema „Decoding the book of life“. Das geschickt gewählte Thema verspricht die neue Dimension: Erstmalig waren die Wissenschaftler in der Lage, Leben in seiner Vollständigkeit und nicht nur Teile davon zu begreifen. Der anfänglichen Euphorie folgte jedoch bald eine gewisse Ernüchterung, denn die Genomsequenz bietet zunächst nur den Bauplan des Lebens, Lebensprozesse können noch nicht allein vom Bauplan abgeleitet werden [2]. Was mit diesem Bauplan dann passiert, wie der Bauplan des Lebens in das wirkliche Leben umgeschrieben wird, sollte auf der Ebene der „functional genomics“ entschieden werden. So befindet die Kontrolle der differenziellen Genexpression darüber, welche Gene wann und mit welcher Intensität exprimiert werden, womit jedes Protein in der benötigten Menge bereitgestellt wird, um am Ende ein komplexes, für jeden Organismus typisches Proteinnetzwerk, ein Kernstück seines Lebens aufzubauen. Ganz entscheidend ist, dass wir heute mithilfe der Multi-Omics-Techniken die Gesamtheit der Transcripte, die Vielfalt nicht codierender RNAs eingeschlossen, der Proteine oder der Metabolite in einer Zelle erfassen können. Um aus dieser Datenfülle neues Wissen abzuleiten, kommt der Bearbeitung und Aufbereitung der ungeheuren Datenmengen, die die Omics- Techniken generieren, durch Bioinformatik und Systembiologie eine Schlüsselstellung zu (Abb. 2).

Abb.2 Von der Genomsequenz uber die Proteine zum Leben – die Genomsequenz ist nur der Bauplan des Lebens, jetzt ist die funktionelle Genomforschung gefragt, den Bauplan des Lebens in das Leben umzuschreiben. Allen voran ist die Proteomics gefragt, das "virtuelle Leben der Gene in das reale der Proteine" umzuschreiben, denn die Proteine, nicht die Gene sind die Spieler des Lebens.

Abb.3 Darstellung des Gesamtproteoms von Staphylococcus aureus A) Die wichtigsten Proteom-Subfraktionen von S. aureus. B) Ein virtuelles 2D-Proteingel: Jeder Punkt reprasentiert ein Protein. Die Lage des Proteins auf dem Gel wird bestimmt durch seine Grose und seine Ladung. C) Ubersicht uber die vorhergesagten und tatsachlich nachgewiesenen Proteine. Abdeckung des Proteoms 76 Prozent. Wenn man berucksichtigt, dass zum gemessenen Zeitpunkt nicht alle Gene exprimiert werden, ist die Abdeckung noch hoher. (mod. nach Becher et al., PloS One 4, 2009, e8176; Hecker et al., Laborwelt 15, 2014, 5)
Was kann die Proteomics zum besseren Verständnis der Pathophysiologie von S. aureus leisten?
Nach Vorlage der Genomsequenz von S. aureus waren wir in der Lage, nahezu die Gesamtheit seiner Proteine – sein Proteininventar – zu identifizieren [4, 5]. Neben bereits bekannten Proteinen wurden dabei wie bei anderen Bakterien auch solche gefunden, die noch niemals beschrieben wurden, solche mit unbekannter Funktion also. Die insgesamt fast 2.000 Proteine wurden von Dörte Becher nach verschiedenen Kriterien geordnet. Zunächst haben wir cytosolische und membranständige Proteine von solchen getrennt, die oberflächenassoziiert aus der Zelle herausragen oder die nach außen transportiert werden (Sekretom). Danach haben wir alle Proteine – soweit bekannt – Funktionseinheiten zugeordnet (Abb. 3 und 4). So konnte z. B. nahezu der gesamte Stoffwechsel, der fast die Hälfte aller Proteine beansprucht, rekonstruiert werden, nicht nur aus der etwas vagen genomischen Voraussage heraus, sondern von den realen Lebensprozessen der Bakterien abgeleitet. Daneben wurden zahlreiche Proteine den grundlegenden Funktionen des Lebens wie der Genexpression einschließlich ihrer Regulation, der Translation und Proteinqualitätskontrolle, der Signaltransduktion und vielen anderen Prozessen zugeordnet. Am Ende wurde auf der Ebene der Proteine das Leben einfacher Organismen in einer bisher kaum gekannten Vollständigkeit abgebildet. Mithilfe der quantitativen Proteomics kann man darüber hinaus auch die Investition für die eben beschriebenen Lebensprozesse berechnen und beispielsweise die Frage beantworten, wie „teuer“ der Zelle die Glykolyse oder die Translation wird (siehe Tabelle 1).
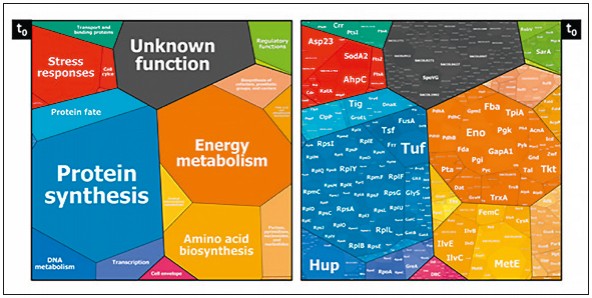
Abb.4 Die Zuordnung der Proteine von S. aureus zu Funktionseinheiten. Die Größe der jeweiligen Flächen ist ein Maß für die vorhandene Menge (nach Bernhardt et al., unveröffentlicht)

Tab.1 Investitionen und Kosten für das „einfache Leben“ von S. aureus (D. Zühlke, J. Bernhardt und S. Fuchs, unveröffentlicht)
Tanz der Proteine und das Leben – Gedanken über die infektionsbiologische Thematik hinaus Die globalen, ausgeklügelten Mechanismen der Genexpressionskontrolle garantieren, dass jedes einzelne Protein in der benötigten Menge und zum rechten Zeitpunkt bereitgestellt wird, wie wir in einer quantitativen Modellstudie zur Beantwortung des Sauerstoffmangels durch S. aureus gezeigt haben, ein unter Infektionsbedingungen sehr häufiges Ereignis im Wirt. Dabei konnten wir bei den durch Sauerstoffmangel massiv induzierten Proteinen nicht nur solche nachweisen, die die Umstellung auf Fermentationsprozesse bewirken (z. B. Lactatdehydrogenase), sondern auch solche unbekannte Funktion, deren detailliertes Studium wichtige Aufschlüsse über bisher unbekannte Mechanismen der Anpassung an das Infektionsgeschehen bietet. Mit dieser Vorlage und Beschreibung des Proteininventars ist ein wichtiger Schritt auf dem Weg vom Genom über das Proteom zum Leben zurückgelegt, Lebensprozesse einfacher Bakterien können in einer Vollständigkeit verfolgt und beschrieben werden, wie wir das vor 20 Jahren noch für undenkbar gehalten haben. Was bleibt auf diesem Wege noch zu tun, was ist der logisch folgende Schritt? Nicht der Proteinmix allein macht das Leben aus, Leben beginnt erst mit dem „geordneten Tanz der Proteine“! Die Herausforderung der kommenden Jahre wird es sein zu verstehen, wie die Lücke von der Bereitstellung des Proteininventars bis zur Zellphysiologie geschlossen wird, wie diese am Ribosom in genau abgestimmter Menge freigelassenen Proteine das eigentliche Leben organisieren. Von Bernd Bukau (Heidelberg) wissen wir, dass sie bereits während der „Geburt am Ribosom“ ihre Partner finden, sie bilden nachfolgend ein dynamisches, durch die Umweltbedingungen gesteuertes hoch sensibles und vermutlich hochgradig geordnetes Proteinnetzwerk aus, das fast alle Lebensprozesse steuert. Die fortgeschrittenen Kenntnisse über das Proteininventar machen S. aureus zu einem gefragten Modellorganismus , der über die infektionsbiologischen Themen hinaus bei der Suche nach der Antwort auf Schrödingers berühmte Frage „What is life?“ hilfreich sein könnte.

Tab.1 Auf dem Weg zum menschlichen Immunproteom von S. aureus – ein Beispiel Die sekretierten Proteine von S. aureus (Stamm 8425) wurden mittels Gelelektrophorese zweidimensional aufgetrennt und erscheinen als orangefarbene Spots. Nach Ubertragung der bakteriellen Proteine auf eine Membran wurde diese mit Serum von 16 Erwachsenen inkubiert und die Bindung von IgG-Antikorpern sichtbar gemacht (blau). Man erkennt deutlich, dass das Immunsystem auf manche Proteine stark, auf andere dagegen schwach oder gar nicht reagiert. Die Gesamtheit der bakteriellen Proteine, die eine Antikorper- oder T-Zellantwort auslosen, bezeichnet man als Immunproteom.
-> hecker@uni-greifswald.de
Literatur Bild: © istockphoto.com| nicolas |
L&M 2 / 2016
Das komplette Heft zum kostenlosen Download finden Sie hier: zum Download Die Autoren:Weitere Artikel online lesen

NewsSchnell und einfach die passende Trennsäule finden
© Text und Bild: Altmann Analytik ZEISS stellt neue Stereomikroskope vor
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen. © Text und Bild: Carl Zeiss Microscopy GmbH |








