


|
Caseine in Frischmilch
Caseine in FrischmilchHPTLC-MS-Imaging von Proteinen und ProteinderivatenIn der Proteinanalytik sind neben den traditionellen Fragestellungen wie der Identifizierung und Quantifizierung von Proteinen Proteinmodifikationen (sogenannte post-translationale Modifikationen – PTM) mittlerweile von besonderem Interesse. Durch diese Modifikationen wird nicht nur die Wirkung spezifischer Proteine determiniert, sondern modifizierte Proteine können zudem als Biomarker in der Physiologie oder als Prozessmarker in der Lebensmittelanalytik herangezogen werden. 
Die aktuelle Analytik beruht auf chromato- graphischen und elektrophoretischen Trennmethoden, die im Hinblick auf Proteinveränderungen oft nur zu unbefriedigenden Ergebnissen führen, da die zugrundeliegenden Reaktionen, die zu diesen PTM führen, sehr komplex sein können und oftmals noch nicht hinreichend charakterisiert sind. Ziel der Kopplung der Dünnschicht- chromatographie (HPTLC) mit der Massen- spektrometrie (MS) ist es, die Vorteile beider Verfahren auszunutzen. Dieser Neo-Proteomics-Ansatz soll es ermöglichen, auch PTM zu identifizieren, die in ihrer Anzahl und Diversität der chemischen Strukturen sehr komplex sein können. Als Beispiel zu nennen sind hier ungerichtete Reaktionen von Proteinen mit anderen Lebensmittelinhaltsstoffen, aber auch solche, die in vivo ablaufen. So finden z.B. zahlreiche Glykierungen von Proteinen statt, die beim Krankheitsbild Diabetes mellitus als Biomarker herangezogen werden können. In diesem Zusammenhang ergeben sich durch die Vorgehensweise des sogenannten HPTLC-MS-Imagings zusätzliche Vorteile: Neben dem direkten Nachweis auf/von der Dünnschichtplatte können durch das Elutionsverhalten Aussagen über ausgewählte physikochemische Eigenschaften der Proteine/Peptide (u.a. Polarität) getroffen und das Molekulargewicht ermittelt werden. Das Imaging ermöglicht den Nachweis von Modifikationen und die Visualisierung der Protein/Peptid-Verteilungen auf der Dünnschichtplatte. Damit können eine schnelle Übersicht und der Vergleich einzelner Proben erreicht werden. Anhand der Analytik von Caseinen aus Frischmilch wurde eine solche methodische Vorgehensweise geprüft. Ziel war der exemplarische Nachweis verschiedener Caseine nach chromatographischer Trennung auf einer Dünnschichtplatte und der anschließenden massenspektrometrischen Kartierung (Imaging) der einzelnen Peptide. Was man wissen muss Der Begriff Proteomics beschreibt das Forschungsgebiet, das sich mit der Analyse der Gesamtheit der Proteine beschäftigt, die von einem Organismus, einem Gewebe oder einer Zelle gebildet werden. Dadurch sollen die Eigenschaften der Proteine und die damit verbundenen biologischen Prozesse besser verstanden werden [1]. Zur Analyse dieser komplexen Fragestellung(en) werden bisher zwei Ansätze verfolgt: die Top-down- oder die Bottom-up-Methode. Beim Bottom-up-Ansatz werden die Proteine vor der massenspektrometrischen Analyse proteolytisch – meistens mithilfe des Enzyms Trypsin – in kleinere Untereinheiten gespalten. Die erhaltenen Peptide werden chromatographisch getrennt, sodass durch die anschließende Massenspektrometrie die Peptidsequenzen und ggf. Modifikationen identifiziert und charakterisiert werden können [2]. Beim Top-down-Ansatz werden hingegen die intakten Proteine voneinander getrennt und anschließend massenspektrometrisch analysiert. Hier werden erst während der Massenspektrometrie durch die Ionisation einzelne kleinere Fragmente erzeugt. Aus dem Spektrum dieser Fragment-Ionen lassen sich schließlich die Proteinsequenz und -modifikationen ermitteln. Eine Trennung und Identifikation gestaltet sich hierbei allerdings wesentlich schwieriger, dafür kann man die komplette Information des Proteins ohne wesentliche Verluste erfassen, was mit dem Bottom-up-Ansatz nicht möglich ist [2]. Die Dünnschichtchromatographie ist ein lange bekanntes Trennverfahren, das aufgrund mangelnder Leistungsfähigkeit bisher vergleichsweise wenig Anwendung in der modernen Hochleistungsanalytik gefunden hat. In Form der High Performance Thin Layer Chromatography (HPTLC) wird diese Methode gegenwärtig aber zunehmend als hochempfindliche, präzise und schnelle Technik mit Anwendungspotenzial in verschiedenen analytischen Bereichen eingesetzt [3]. Durch Hochleistungsgeräte für die Auftragung, Entwicklung und Dokumentation sowie die Verbesserung der Sorbentien konnten eine Steigerung der Trennleistung und eine Erhöhung der Präzision sowie der Reproduzierbarkeit erreicht werden. Ein großer Vorteil dieses Verfahrens besteht in der Möglichkeit der simultanen Analyse mehrerer Proben sowie der Mehrfach- und mehrdimensionalen Entwicklung. Weitere Vorteile sind die vielfältigen Detektions- und Kopplungsmöglichkeiten, insbesondere die Kopplung der HPTLC mit der Matrix Assisted Laser Desorption/Ionisation Mass Spectrometry (HPTLC-MALDI-MS), der Electrospray-Ionisation Mass Spectrometry (HPTLC-ESI-MS) oder der Desorption Electrospray Ionisation Mass Spectrometry (HPTLC-DESI-MS) [4, 5, 6]. Eine neue Entwicklung sind spezielle MS-grade Platten für die Kopplung der TLC mit der Massenspektrometrie. Damit werden eine hohe Empfindlichkeit, geringere Hintergrundsignale und dadurch sehr gute Signal/Rausch-Verhältnisse erreicht [7]. Wie man vorgeht Der Arbeitsablauf des HPTLC-MALDI-Imagings gliedert sich in drei wesentliche Schritte: (I) Probenvorbereitung und chromatographische Trennung, (II) die massenspektrometrische Messung und (III) das eigentliche Imaging – die Darstellung der massenspektrometrischen Daten und deren Auswertung (Abb. 1).

Abb.1 Schematischer Ablauf der Untersuchung einer Probe durch die 2D-HPTLC-MALDI-Massenspektrometrie.
I) Probenvorbereitung und Dünnschichtchromatographie Die Caseine können aufgrund ihrer Löslichkeit bei pH 4,6 leicht von der Milch und Molkenproteinen abgetrennt werden, nachdem störendes Fett durch Zentrifugation entfernt wurde. Es existieren diverse Caseine in der Milch (u.a. auch abhängig von der Milchherkunft, „Kuh vs. Schaf“), wobei das aS1-Casein, das aS2-Casein, das ß-Casein und das k-Casein die bekanntesten Vertreter sind. Für das verwendete analytische Verfahren werden die Caseine ohne vorherige Trennung proteolytisch durch Trypsin gespalten. Die Trennung der resultierenden Peptide erfolgt auf der Dünnschichtplatte (hier: eine HPTLC Kieselgel 60 F254 MS-grade für MALDI, Merck KGaA, Darmstadt, Deutschland). Die Trennung kann nach Auftragung der Probe in einer Bande in einer Dimension oder nach punktförmiger Auftragung in zwei Dimensionen stattfinden (Abb.2). Für die zweidimensionale Trennung wird die HPTLC-Platte zuerst in der ersten Dimension mit 2-Butanol/Pyridin/Ammoniak/Wasser als mobiler Phase entwickelt. Nach der vollständigen Trocknung der Platte wird diese um 90° gedreht und anschließend mit 2-Butanol/Pyridin/Essigsäure/Wasser als mobiler Phase in der zweiten Dimension entwickelt. Die beiden mobilen Phasen unterscheiden sich abseits von den Lösungsmittelverhältnissen hauptsächlich in der Wahl eines modifizierenden Agens („Modifier“; i.d.R. Essigsäure, Ammoniak, Tetrahydrofuran etc.). In der ersten Dimension wird Ammoniak für einen basischen pH-Wert und in der zweiten Dimension Essigsäure für einen sauren pH-Wert verwendet. Die Trennung in der Dünnschichtchromatographie ist ein komplexes Zusammenspiel aus der Interaktion der Analyten mit der stationären und der mobilen Phase. Durch die unterschiedlichen pH-Werte werden die Ladung und damit auch die Polarität der Peptide beeinflusst, wodurch unterschiedliche Retentionsfaktoren (Rf-Werte) erreicht werden. Dies bedeutet unterschiedliche Wanderungsgeschwindigkeiten eines Peptides und damit unterscheidbare Entfernungen des Peptids vom Trennungsursprung, der abhängig vom zugesetzten Modifier ist. Die Verwendung von parallel entwickelten Dünnschichtplatten erlaubt dabei den direkten Vergleich zwischen den Detektionsmethoden (Abb.1). Gerade im Bereich der Protein- und Peptidanalytik können traditionelle Farbreagenzien erste Informationen über die Eigenschaften liefern, z.T. auch schon über die chemische Struktur. Dabei kommen primär Farbreagenzien wie Ninhydrin oder Fluorescamin zum Einsatz, die entsprechend ihrer Interaktion mit den Analyten auf der Dünnschichtplatte den Nachweis ihrer Präsenz, aber auch ihrer Verteilung auf der Platte ermöglichen (Abb.2). Die Vielzahl möglicher Derivatisierungsreagenzien kann hier eindeutig als Vorteil der Dünnschichtchromatographie angesehen werden.
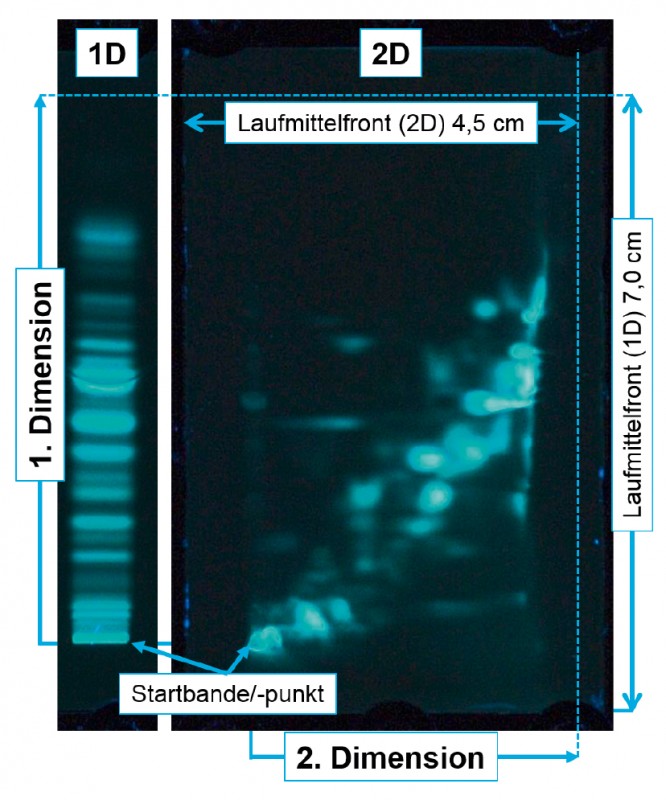
Abb.2 Peptide des tryptischen Verdaus von Milchproteinen nach ein- und zweidimensionaler Entwicklung. Derivatisiert mit Fluorescamin und visualisiert unter UV-Licht (366?nm).
II) Kopplung der Dünnschichtchromatographie mit der MALDI-Massenspektrometrie Im Fall der MALDI-TOF-MS-Kopplung wird nach der chromatographischen Trennung die Oberfläche der HPTLC-Platte komplett mit einer geeigneten MALDI-Matrix beschichtet. Dies kann durch Tauchen oder Aufsprühen erfolgen. Anschließend wird die Platte für die Messung in einen (kommerziell erhältlichen) Adapter eingebracht und zur Messung in das Massenspektrometer eingesetzt (Abb.3). Die vollautomatisierte Messung wird im Falle einer 1D-Trennung bahnweise durchgeführt und im Falle einer 2D-Trennung, indem der Laser sich anhand eines Rasters von Quadraten definierter Kantenlänge über den Messbereich der Platte bewegt. In jedem Rasterpunkt wird dabei ein Massenspektrum aufgenommen. Aus der Gesamtinformation aller Rasterpunkte wird dann ein massenspektrometrisches Image erstellt, in dem die detektierten Massen abhängig von ihrer Intensität in den einzelnen Spektren farbig dargestellt werden können (Abb.4).

Abb.3 Schematische Darstellung der Laserdesorption auf der TLC-Platte unter Anwendung eines kommerziellen HPTLC-MALDI-MS-Adapters.

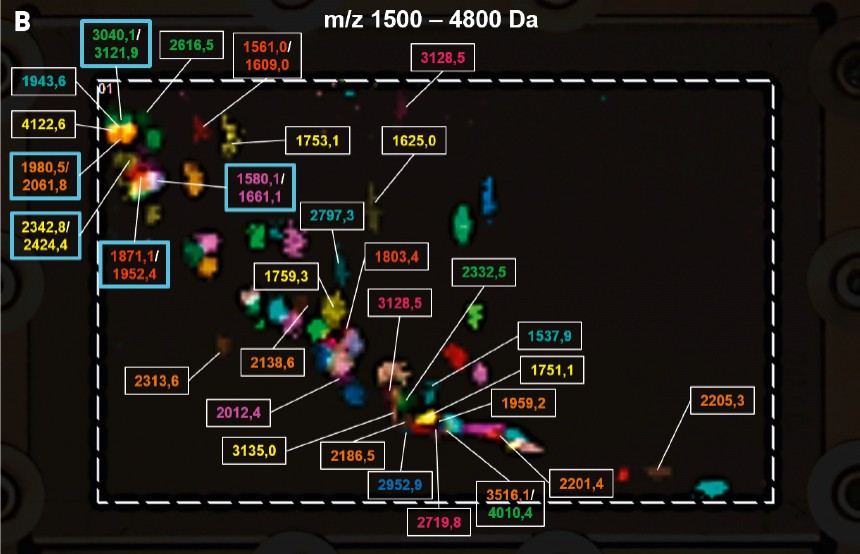
Abb.4 A und B Darstellung des zweidimensionalen HPTLC-MALDI-Imagings mit Beschriftung der Massen aller 89 detektierten Caseinpeptide. (A) Massenbereich 600 – 1500 Da; (B) Massenbereich 1500 – 4800 Da.
III) Auswertung und Ergebnisse Durch den Vergleich des MALDI-Images mit dem „Image“ einer traditionell angefärbten Platte können Peptide, die zuvor nur aufgrund der Fluoreszenzfärbung oder nach Derivatisierung detektiert wurden, anhand ihrer Masse identifiziert werden. Die in der Dünnschichtchromatographie übliche Verwendung von Standards während der Trennung ist dadurch nicht mehr absolut nötig, da jede Bande (1D), beziehungsweise jeder Spot (2D) in den meisten Fällen eindeutig durch die Massenspektrometrie identifiziert werden kann. Bei der exemplarischen Untersuchung der aus Frischmilch gewonnenen Caseine konnten mit dieser Vorgehensweise auf einer einzelnen Platte 89 Peptide nach 2D-Entwicklung nachgewiesen werden. Aufgrund dieser großen Anzahl bzw. für eine erleichterte Evaluierung erfolgt die Beschriftung (Annotation) in zwei arbiträren Massenbereichen für ein und dieselbe Platte (hier: 600–1500 Da und 1500–4800 Da in Abb.4A und B). Dadurch kann auch die Verteilung der Peptide auf der Platte gut visualisiert werden. Das Beispiel der Caseinpeptide zeigt eine Unabhängigkeit der Trennung von der Peptidmasse. Im Allgemeinen können große und kleine Peptide über den gesamten Bereich der Trennung detektiert werden, da diese wie oben beschrieben vornehmlich durch die Polarität determiniert ist und durch die chromatographischen Bedingungen selektiv beeinflusst werden kann. Zwischen den Peptiden, der stationären Phase und der mobilen Phase bestehen multiple Wechselwirkungen, welche stark von der Aminosäuresequenz des einzelnen Peptides abhängig sind. Die einzelnen Aminosäuren bestimmen dabei unter anderem auch die Ladung des Peptids, die durch den pH-Wert (der Eluenten) beeinflusst werden kann. Dadurch können auch Peptide mit einem sehr geringen Massenunterschied von 1 Da unterschiedliche Rf-Werte aufweisen (durch „#“ markierte Beispiele in Abb.5).
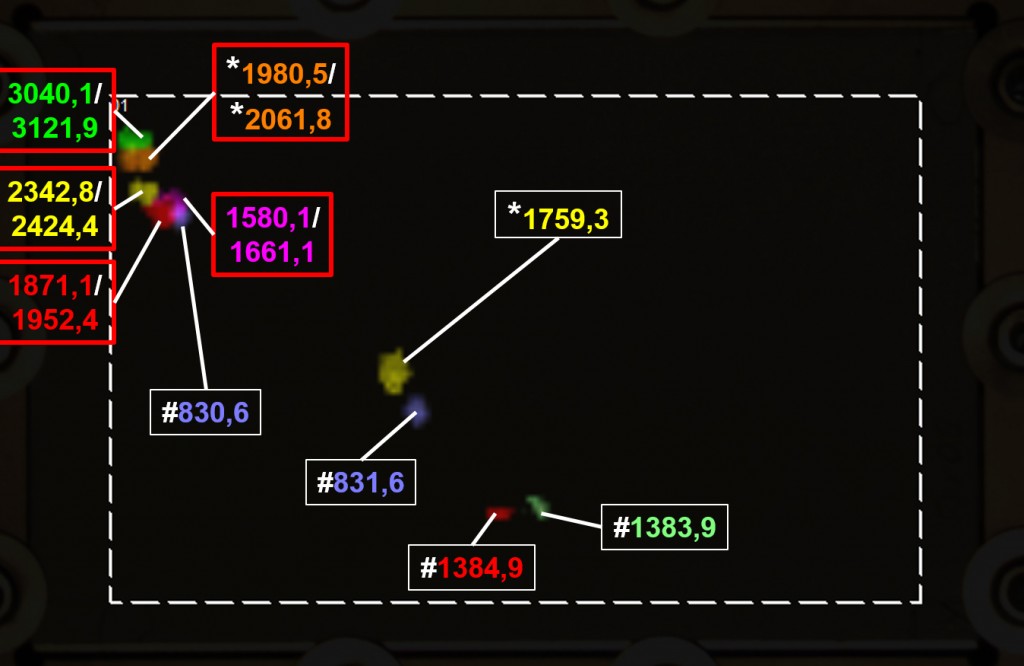
Abb.5 Darstellung einer reduzierten Anzahl von Peptiden des 2D-HPTLC-MALDI-Imagings aus Abbildung 4. Phosphorylierte Peptide sind mit roten Rahmen, vollständig durch Fragmentierung sequenzierte Peptide durch Sternchen („*“) und Peptide mit geringer Massendifferenz aber unterschiedlicher Position durch Rauten („#“) markiert.
Phosphorylierte Peptide können oftmals nur schwer voneinander getrennt werden und weisen unter den meisten üblichen Chromatographiebedingungen nur sehr geringe Rf-Werte auf. Allerdings erlaubt hier die Kopplung mit der Massenspektrometrie trotz der geringen Trennung die Identifizierung der Phosphopeptide anhand der Messung des Verlusts der Phosphorylierung (Neutralverlust 80 Da) in den sich anschließenden MS/MS-Untersuchungen (durch einen roten Rahmen markierte Beispiele in Abb.5). Phosphorylierungen sind in der Regel PTM der Aminosäuren Serin oder Threonin. Über die Fragmentierung der Peptide in der MS/MS-Analytik ist die Aufklärung der Aminosäuresequenz und damit der modifizierten Aminosäure eines Peptides möglich. Ein für die Sequenzierung eines phosphorylierten Peptides anschauliches Beispiel ist das Peptid-Paar mit 1980,5 Da und 2061,8 Da (mit Sternchen ‚*‘ markiert in Abb.5). Diese korrespondieren einerseits mit dem unmodifizierten Peptid FQSEEQQQTEDELQDK mit einer Masse von 1981,9 Da und dem phosphorylierten Peptid FQS*EEQQQTEDELQDK mit einer Masse von 2061,9 Da andererseits. Ein weiteres Beispiel für ein vollständig sequenziertes Peptid wurde mit der Masse 1759,3 Da gemessen. Dieses korrespondiert mit dem Peptid HQGLPQEVLNENLLR mit einer theoretischen Masse von 1759,9 Da. Fazit – von der Frischmilch zu Zucker und Lipiden Die zahlreichen Freiheitsgrade der verschiedenen Trennsysteme der HPTLC ermöglichen eine vielseitige und umfassende Analyse zur Identifizierung und Charakterisierung verschiedener Proteine/Peptide. In Kombination mit der Massenspektrometrie und dem Imaging können neben einer einfachen Bestimmung des Molekulargewichts der einzelnen Peptide und verschiedenen Modifikationen Informationen über die Proteinsequenz direkt von der HPTLC-Platte „auf einen Blick“ erhalten werden. Beim TLC-MALDI-Imaging, beispielhaft angewendet für die Analyse von Frischmilch, konnten die vier wichtigsten Kuhmilch-Caseine einfach und sicher durch wenige spezifische Peptide nachgewiesen werden. Das Verfahren lässt sich auch leicht auf andere Proteine und Peptide, aber auch vollkommen andere Analyten wie Lipide oder Zucker anwenden. Im Hinblick auf physiologische Veränderungen, d.h. Modifikationen von Proteinen, aber auch Veränderungen während der Be- und Verarbeitung von Lebensmitteln können damit auf vielfältige Weise genauer betrachtet werden.
Literatur Foto: © istockphoto.com| Vizerskaya |
L&M 8 / 2015
Das komplette Heft zum kostenlosen Download finden Sie hier: zum Download Die Autoren:Weitere Artikel online lesen


NewsSchnell und einfach die passende Trennsäule finden
© Text und Bild: Altmann Analytik ZEISS stellt neue Stereomikroskope vor
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen. © Text und Bild: Carl Zeiss Microscopy GmbH |









