


|
Lokalisationsmikroskopie – neue Markierungsstrategien für bessere Auflösung
Lokalisationsmikroskopie – neue Markierungsstrategien für bessere AuflösungLicht am richtigen OrtMithilfe der Lokalisationsmikroskopie können zelluläre Strukturen unterhalb von 200 nm mit hohem Kontrast und hoher räumlicher Auflösung dargestellt werden. Dazu muss die Zielstruktur mit photoschaltbaren Farbstoffen markiert werden. Dies kann mithilfe der Immunfluoreszenz erfolgen oder man lässt die Zelle selbst photoschaltbare fluoreszierende Proteine exprimieren. Alternativ kann die Nanoinjektion für intrazelluläre Färbungen eingesetzt werden. Hochauflösende Fluoreszenzmikroskopie 
Die hochauflösende Fluoreszenzmikroskopie ermöglicht es, zelluläres Leben mit bisher nicht dagewesener räumlicher Auflösung zu erforschen. Die klassische Auflösungsgrenze kann dabei über die zeitliche Trennung der Fluoreszenzsignale umgangen werden. Während deterministische Methoden wie STED (Stimulated Emission Depletion) und SIM (Structured Illumination Microscopy) mit modifizierten Laseranregungsmustern arbeiten, beruht die Lokalisationsmikroskopie auf der stochastischen Detektion einzelner Moleküle und deren präzisen Positionsbestimmung. Zu den ersten Umsetzungen dieses Prinzips zählen PALM (Photoactivated Localization Microscopy) [1] und STORM (STochastic Optical Reconstruction Microscopy) [2]. Während bei PALM photoaktivierbare fluoreszierende Proteine zum Einsatz kommen, benutzt STORM als photoschaltbare Einheit ein Paar aus organischen Farbstoffmolekülen. Die Methode dSTORM (direct STORM) ist eine Weiterentwicklung von STORM, bei der konventionelle Farbstoffe mithilfe von chemischen Puffern als Photoschalter nutzbar gemacht werden können [3]. Das Prinzip der Lokalisationsmikroskopie Bei der Lokalisationsmikroskopie wird eine Struktur mit photoschaltbaren Farbstoffen markiert, die als konventionelles Fluoreszenzbild am Mikroskop sichtbar gemacht werden kann (Abb.1). Um einen dSTORM-Film aufzunehmen, wird bei der Aufnahme die Mehrzahl der Farbstoffe in einen nicht fluoreszierenden Dunkelzustand versetzt und nur einige wenige Farbstoffe werden zufällig in den fluoreszierenden An-Zustand befördert. Über einen Zeitraum von wenigen Minuten wird mithilfe von lichtempfindlichen Kameras ein Film aufgenommen, der aus mehreren Tausend Einzelbildern besteht. Das Emissionsmuster räumlich isolierter Farbstoffe kann nun mathematisch approximiert werden und so auf die Position des Moleküls geschlossen werden (Lokalisation). Die Lokalisation ist umso genauer, je mehr Photonen detektiert werden. Aus allen Lokalisationen kann ein hochaufgelöstes Bild rekonstruiert werden, dessen Auflösung im Bereich von 20 nm und weniger liegen kann.

Abb.1 Prinzip der Lokalisationsmikroskopie. Zelluläre Strukturen werden mit photoschaltbaren Fluorophoren markiert, z.B. mithilfe von Antikörpern (primärer Antikörper in Hellgrau, farbstoffmarkierter sekundärer Antikörper in Dunkelgrau). Unter Laseranregung wird auf dem Mikroskop die Fluoreszenz der Farbstoffe mithilfe von lichtempfindlichen Kameras eingefangen. Der Großteil der Farbstoffe wird in einen nicht fluoreszierenden Aus-Zustand befördert. Nur einige wenige Farbstoffe werden stochastisch in den fluoreszierenden An-Zustand geschaltet. Der Schaltprozess wird während der Filmaufnahme mehrere Tausend Male wiederholt. Einzelne Farbstoffe der Einzelbilder können mathematisch nanometergenau lokalisiert werden. Schlussendlich wird ein hochaufgelöstes Bild aus allen Lokalisationen rekonstruiert.
Effiziente und spezifische Markierung Die Markierung der Probe stellt einen zentralen Bereich der Lokalisationsmikroskopie dar. Dies kann im Fall von dSTORM mithilfe der Immunmarkierung oder im Fall von PALM über eine Transfektion geschehen. Bei der Immunmarkierung werden Zielproteine mit Farbstoff-modifizierten Antikörpern gefärbt, oft kommen auch zwei Antikörper – ein erster ist gegen das Zielprotein gerichtet, ein zweiter gegen den ersten – zum Einsatz, während bei der Transfektion Zellen gentechnisch dazu gebracht werden, ein an das Zielprotein gekoppeltes fluoreszierendes Protein (FP) zu exprimieren. Idealerweise ist die Markierung hoch spezifisch und vollständig. In der Realität können allerdings Epitope durch die chemische Fixierung der Proben verloren gehen und nicht alle Epitope sind für Antikörper zugänglich, sodass nicht garantiert werden kann, dass alle Proteine bei der Markierung mit Antikörper erfasst werden können. Aber auch bei stabil transfizierten Zellen, bei denen alle Zielmoleküle zusammen mit FPs exprimiert werden, ist die anschließende Detektion unvollständig, da nicht alle FPs maturieren und dementsprechend fluoreszieren können. Informationsgewinn durch Mittelung Unvollständige Markierung kann nicht vermieden werden. Allerdings können symmetrische Strukturen, die unvollständig markiert sind, aber innerhalb der zu untersuchenden Probe mehrfach vorkommen, zu einem perfekt aufgelösten Abbild der Struktur gemittelt werden. Diese Methode stammt aus der Elektronenmikroskopie und kann auf Lokalisationsdaten angewendet werden. Dies ist in Abbildung 2 am Beispiel des Kernporenkomplexes dargestellt. Das Protein gp210 ordnet sich innerhalb des Komplexes mit einer achtfachen Symmetrie an, welche auch schon an einzelnen hochaufgelösten Strukturen im rekonstruierten dSTORM Bild erkannt werden kann. Werden einzelne Kernporen nun isoliert und deren Lokalisationen ausgerichtet und aufsummiert, kann aus allen Lokalisationen ein Bild gemittelt werden (Abb.2). Die dargestellte Struktur kann nun sehr genau geometrisch charakterisiert werden [4]. Dieses Verfahren wurde auch an hochaufgelösten Herpesviren angewendet. So konnte die radiale Anordnung von viralen Proteinen zwischen Kapsid und Virenhülle durch Mittelung vieler hochaufgelöster Virenpartikel sehr präzise bestimmt werden [5].
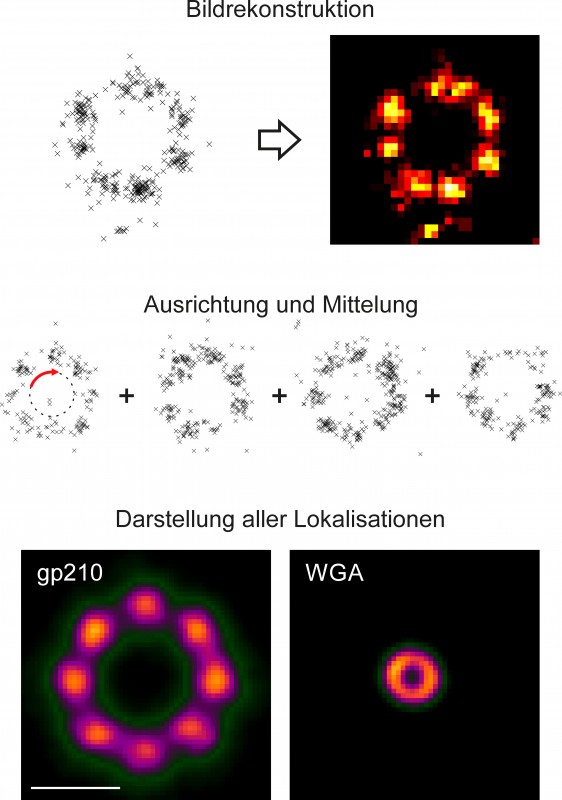
Abb.2 Mittelung von Einzelmolekül-Lokalisationen. Oben: Lokalisationsmuster des Kernporenproteins gp210 und rekonstruiertes dSTORM-Bild. Mitte: Zur Mittelung werden die Lokalisationsmuster einzelner Kernporen ausgerichtet und aufsummiert. Unten: Gemittelte Bilder des Kernporenproteins gp210 (links, 164?nm Durchmesser) und des zentralen Kanals (rechts, 41?nm Durchmesser) [4]. Maßstab 100?nm
Höhere Auflösung durch kleinere Labels Neben Antikörpern steht zur Färbung zellulärer Strukturen eine Reihe von Alternativen zur Verfügung. So können farbstoffmodifizierte Toxine benutzt werden (z.B. Phalloidin oder Paclitaxel). Ferner erlauben es sogenannte Tags (Halo, SNAP, CLIP), Zellen mit organischen Farbstoffen zu markieren. Diese Tags haben den Vorteil, dass sie sehr klein sind (kleiner als ein GFP-Molekül, s. Abb.3); die Zellen müssen aber zuvor transfiziert werden, damit die Tags exprimiert werden können. Zur Visualisierung des zentralen Kanals des Kernporenkomplexes ist Weizenkeim-Agglutinin (WGA) verwendet worden, das mit 38kDa als Label gerade klein genug ist, um den Kanal auflösen zu können (Abb.2) [4]. Eine weitere Alternative stellen Einzeldomänen-Antikörper (Nanobodies) dar, die aus Kamelen und Haien gewonnen werden. Der Vorteil von Nanobodies ist neben hoher Spezifität eine ebenfalls reduzierte Größe gegenüber Antikörpern (Abb.3). Während bei der indirekten Immunfluoreszenz zwei etwa 10nm große Antikörper (~150kDa) das Zielprotein markieren, kann mithilfe der ca. 2–4nm kleinen Nanobodies (~15kDa) eine bessere Auflösung erreicht werden [6].
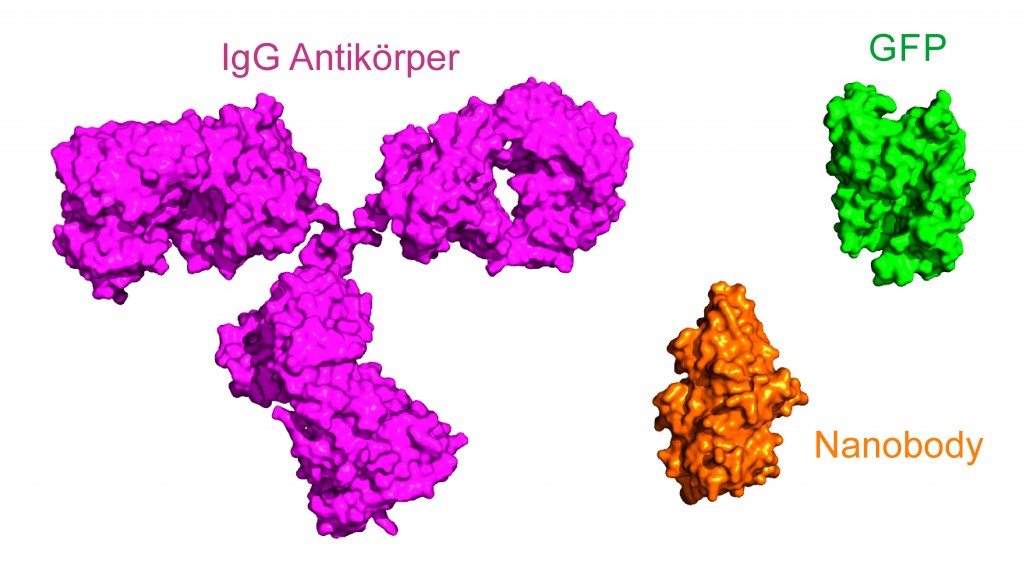
Abb.3 IgG Antikörper, GFP und Nanobody (VHH-Domäne) im Größenvergleich. GFP: 2,4nm × 4,2nm
Nanoinjektion Eine weitere Möglichkeit der Fluoreszenzmarkierung ist die neu entwickelte Nanoinjektion [7], die mithilfe einer 100nm schmalen Nanopipette Moleküle gezielt in einzelne Zellen bringt (Abb.4). Dies geschieht über elektrophoretische Kräfte. Mit dieser Technik, die für die Zelle wesentlich schonender als die klassische Mikroinjektion ist, kann zusätzlich die Markierungsdichte aktiv gesteuert und so eine vollständige Markierung der Zielstruktur innerhalb der Zelle sichergestellt werden. Weiterhin umgeht diese Methode die Zellmembran als natürliche Schranke nichtzellpermeabler Fluoreszenzfarbstoffe und erlaubt so den Einsatz aller Photoschalter in lebenden Zellen. Dabei ist die Annäherung an die Zelle so präzise, dass sogar Farbstoffe in einzelne Kompartimente wie den Zellkern direkt injiziert werden können. Die Farbstoffe können auch gebunden an DNA, Toxine und sogar Antikörper injiziert werden. Mithilfe der Nanoinjektion können Färbeprozesse live beobachtet und anschließend mittels Lokalisationsmikroskopie hochaufgelöst werden.
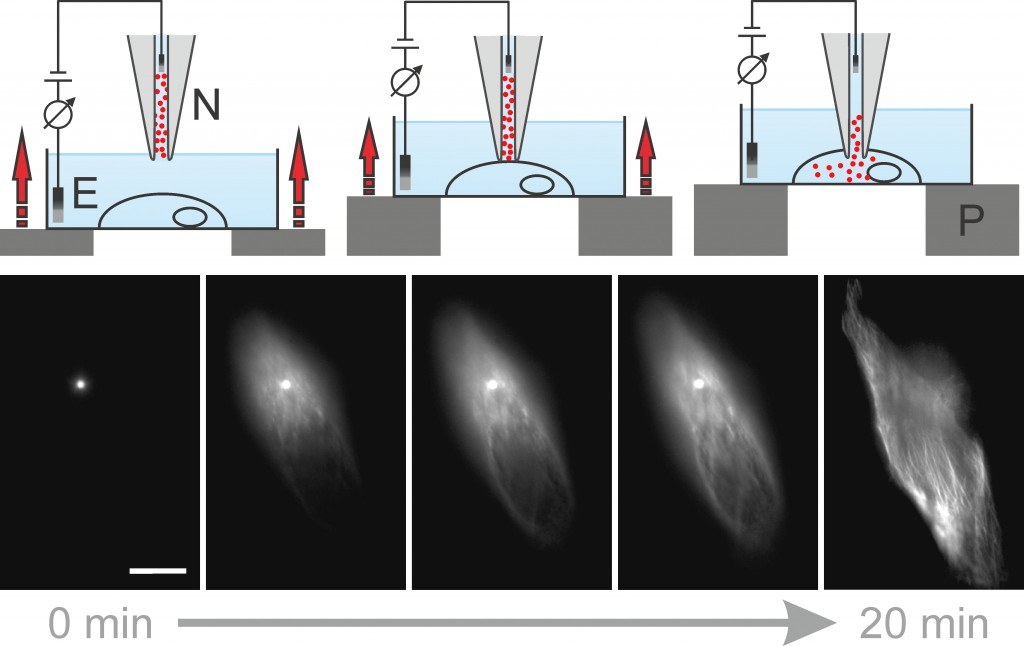
Abb.4 Prinzip der Nanoinjektion. Oben: Nachdem die Nanopipette (N) mit Farbstoffmolekülen gefüllt und manuell ca. 20µm über der Zelle platziert ist, beginnt eine computergesteuerte Annäherung. Ein Piezotisch (P) hebt die Zelle langsam gegen die feststehende Nanopipette. Gleichzeitig ist ein Ionenstrom, der durch die Nanopipette fließt, zwischen den Elektroden (E) messbar. Kommt die Nanopipette mit der Zellmembran in Berührung, so wird dies im Ionenstrom sichtbar. Nachdem die Zellmembran durchstoßen wurde, können die Farbstoffmoleküle über ein angelegtes Potential von der Nanopipette in die Zelle übergehen. Unten: Zelle während der Nanoinjektion von Oregon Green modifiziertem Paclitaxel. Die Moleküle binden – ausgehend von der Nanopipette (sichtbar als heller Punkt) – an die Tubulinstruktur der lebenden Zelle und machen diese unter Laseranregung sichtbar [7]. Maßstab 5µm
Zukünftig werden Markierungsstrategien weiter verbessert werden müssen, um für verschiedenste Zielproteine hochspezifische Färbungen erreichen zu können. Ein Großteil der kommerziell erhältlichen Antikörper ist oft nicht für die Immunfluoreszenz und Lokalisationsmikroskopie geeignet, sodass viele Antikörper von Arbeitsgruppen über Jahre hin selbst optimiert werden müssen.
Literatur: Foto: © fotolia.com| silvae, istockphoto| Eraxion |
L&M 5 / 2015
Das komplette Heft zum kostenlosen Download finden Sie hier: zum Download Die Autoren:Weitere Artikel online lesen


NewsSchnell und einfach die passende Trennsäule finden
© Text und Bild: Altmann Analytik ZEISS stellt neue Stereomikroskope vor
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen. © Text und Bild: Carl Zeiss Microscopy GmbH |







