

|
Chemie
 >
Vom Chemiker zum Biotech-Entrepreneur
>
Vom Chemiker zum Biotech-Entrepreneur
Vom Chemiker zum Biotech-EntrepreneurÜber die Anfänge des Protein-Engineering – neue Wege zu Biomolekülen und deren AnwendungenAls ich im April 1980 mein Ingenieurstudium der Chemie an der damaligen Technischen Hochschule Darmstadt antrat, wäre mir nicht der Gedanke in den Sinn gekommen, dass ich einmal zwei erfolgreiche Biotech-Unternehmen gründen würde, von denen eines sogar den Sprung an die US-Börse Nasdaq schafft. 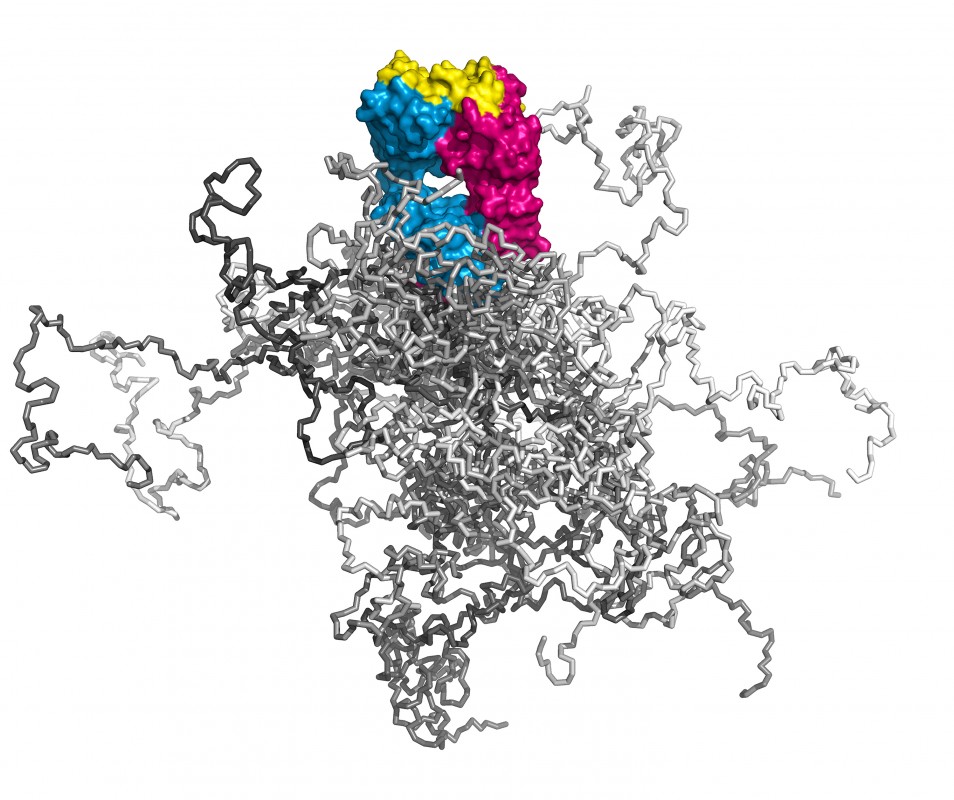
Ein Faible für die Chemie entwickelte sich bei mir bereits in früher Jugend, wo ich gerne mit Fläschchen aus dem Fundus meiner Großmutter im Waschbecken spielte. Im Konfirmandenunterricht lernte ich einen Gleichgesinnten kennen, der mich auf eine Bibliothek in unserem Wiesbadener Stadtteil aufmerksam machte, wo man Chemie-Experimentalbücher ausleihen konnte. Noch bevor der Chemieunterricht in der Schule begann, richteten wir uns in der Waschküche meines Elternhauses ein kleines Labor ein, in dem wir durchaus spektakuläre chemische Versuche veranstalteten. Dass ich schließlich innerhalb des Rhein-Main-Gebietes gerade an der TH Darmstadt mein Studium antrat, war vor allem der gymnasialen Oberstufenreform zu verdanken, die mir ein um ein halbes Jahr vorgezogenes Abitur gestattete, wobei nur dort der Studienbeginn zum Sommersemester möglich war. Neugierde für Biochemie Rückblickend habe ich von meinem Studium mit den vier traditionellen Säulen anorganische, organische, physikalische und technische Chemie sehr profitiert. Insbesondere in den letzten beiden Fächern habe ich an der TH Darmstadt eine selten fundierte fachliche Ausbildung erhalten, die mir auch in der Biochemie später von großem Nutzen gewesen ist. Letztere spielte im regulären Chemiestudium dort allerdings kaum eine Rolle, auch wenn wir mit dem Biochemiker Prof. Hans Günter Gassen einen renommierten Vertreter in unserem Fachbereich hatten – der aber bloß freiwillige Spezialveranstaltungen im fortgeschrittenen Hauptstudium anbot. Mein frühes Interesse an der Biochemie wurde daher eher zufällig beim Studium der Zeitschrift „Spektrum der Wissenschaft“ sowie anhand eines Artikels über gentechnisch hergestelltes Interferon in der „Chemie in unserer Zeit“ [1] geweckt. Als ich mich – dank eines Stipendiums der Studienstiftung des deutschen Volkes mit größerer finanzieller Flexibilität ausgestattet – nach dem Vordiplom für einen Wechsel an eine ferner gelegene deutsche Universität interessierte, boten weder Tübingen noch Heidelberg die von mir angestrebte Schwerpunktsetzung auf die Biochemie innerhalb eines Chemie-Hauptstudiums; möglich war dies jedoch an der Ludwig-Maximilians-Universität München. Dort hörte ich dann also die Biochemie-Grundvorlesung bei Prof. Guido Hartmann, und ich absolvierte das von Prof. Ernst-Ludwig Winnacker organisierte Grundpraktikum im Keller des damaligen Institutsgebäudes in der Karlstraße. Allerdings war dieser recht klassisch ausgelegte Ausbildungsabschnitt (Enzymisolierung aus Gewebehomogenaten, Messungen an alten Analog-Photometern usw.) nicht das, was ich erwartet hatte, und auch das seinerzeit primär auf Faktenwissen ausgerichtete Chemiestudium in München traf nicht ganz meinen Geschmack. Ich entschied mich daher für die Rückkehr an die TH Darmstadt, wo ich zudem ein Interesse an der Computer-Chemie entwickelte und schließlich meine Diplomarbeit bei Prof. Jürgen Brickmann über die Molekulardynamik-Simulation an einem biologischen Ionenkanal abschloss [2]. Ein heißes Thema: Protein-Engineering Während der letzten beiden Semester nahm ich aber noch die Gelegenheit zu einem Ferienpraktikum am Max-Planck-Institut für Biochemie in Martinsried wahr, um das ich mich während meines Studiums in München beworben hatte. Dieses Praktikum eröffnete mir erstmals den Einblick in aktuelle Methoden der Biochemie und Gentechnik einschließlich des Zugangs zu modernen Forschungsinstrumenten, war allerdings für mich zu molekularbiologisch geprägt. Anstoß für die Ausrichtung meines wissenschaftlichen Werdegangs bot dagegen kurz darauf ein Übersichtsartikel von Prof. Alan Fersht und Kollegen über „Protein-Engineering“ in der Fachzeitschrift „Angewandte Chemie“ [3]. In diesem Beitrag wurde das sich im Jahr 1984 erst im Entstehen befindende Forschungsgebiet vorgestellt und es wurde geschildert, wie mithilfe gentechnisch eingeführter Mutationen einzelne Aminosäuren in Enzymen zielgerichtet ausgetauscht werden können, um so Struktur-Funktionsbeziehungen zu studieren. Dieser konstruktive biochemische Denkansatz entsprach dem Konzept der präparativen Synthese neuer Verbindungen, das ich während meines Chemiestudiums kennengelernt hatte. Ich begab mich daher auf die Suche nach einem geeigneten Labor, wo ich diese Art Wissenschaft in meiner Doktorarbeit praktizieren könnte. Hierbei stieß ich am neu gegründeten, von E.-L. Winnacker geleiteten GenZentrum der LMU München, das damals noch in einem Flügel des Martinsrieder MPI untergebracht war, auf die im Aufbau befindliche Arbeitsgruppe des aus den USA zurückgekehrten Nachwuchswissenschaftlers Dr. Andreas Plückthun. Er hatte die Idee, Protein-Engineering mit Antikörpern zu betreiben, und zwar mit dem Schwerpunkt, biokatalytische Aktivität zu generieren – eine damals von der US-Westküste kommende besonders aktuelle Fragestellung. Die Thematik erschien mir spannend und es gelang mir damit, ein Kekulé-Stipendium des Fonds der Chemischen Industrie zu erlangen. Allerdings wurde uns erst während der aktiven Auseinandersetzung mit diesem Projekt klar, dass zuvor eine wesentliche Hürde zu meistern war: Es war nämlich noch keine Methode bekannt, wie man Antikörper (oder zumindest deren funktionelle Fragmente) in einem gut etablierten Expressionsorganismus, wie z.B. Escherichia coli, gentechnisch herstellen könnte. Dieser Aufgabe widmeten sich also für die kommenden Jahre in unserem Labor drei Doktoranden auf unterschiedlichen Pfaden, darunter mein Weggefährte Rudi Glockshuber (heute Prof. an der ETH Zürich). Die Idee für die von mir schließlich verfolgte Expressionsstrategie kam mir beim Studium des gerade neu erschienenen Lehrbuchs „Immunology“ von Prof. Ivan Roitt und Kollegen, das mir der Leiter der Nachbararbeitsgruppe Dr. Thomas Hünig empfohlen hatte. Darin wurde sehr anschaulich die Biosynthese von Antikörpern in ihren natürlichen Wirtszellen, den immunologischen Plasmazellen, beschrieben, wobei vor allem die initiale Sekretion der Immunglobulinketten in das Lumen des endoplasmatischen Retikulums und die dort stattfindende Proteinfaltung unter oxidativer Ausbildung der Disulfidbrücken betont wurden. Mir drängte sich unmittelbar die Vorstellung auf, diese Vorgänge auf die periplasmatische Sekretion in E. coli zu übertragen. Erfahrung im Umgang mit bakteriellen Signalsequenzen hatte ich auf Anregung von A. Plückthun bereits gesammelt; was noch fehlte, war ein gangbares Konzept für die gleichzeitige Expession von leichter und schwerer Immunglobulinkette in derselben Bakterienzelle. Inspirierend hierfür war ein Vortrag von Prof. Ralf Mattes, damals im Pharmaunternehmen Boehringer Mannheim (heute Roche Diagnostics) tätig, in dem er über Versuche zur gleichzeitigen Produktion der beiden Antikörperketten in E. coli berichtete. Allerdings wurden dabei – im Einklang mit der damals üblichen Vorgehensweise – die ursprünglichen Signalsequenzen dieser Polypeptidketten entfernt, um das funktionelle Protein später erst durch Rückfaltung in vitro zu rekonstituieren.
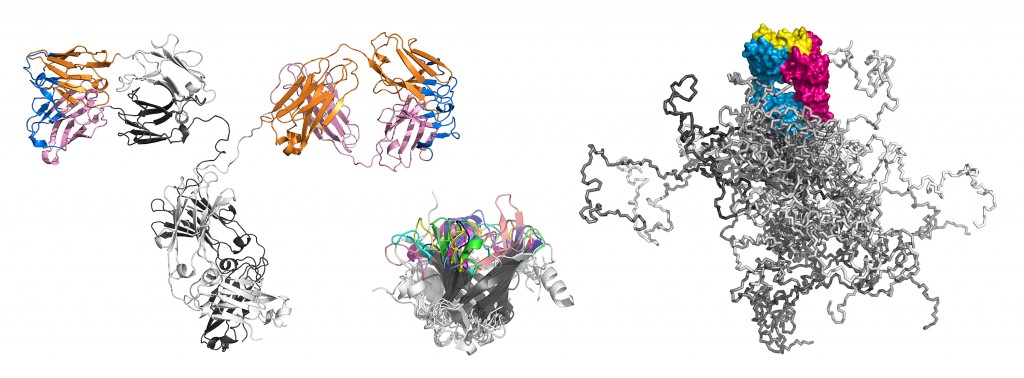
Illustration dreier Molekülformate im Mittelpunkt der Technologien, über deren Entwicklung hier berichtet wird. Links: Ein kompletter Antikörper mit dem farblich hervorgehobenen Fv- und Fab-Fragment; beide tragen die Antigenbindungsstelle und lassen sich mit sehr guten Ausbeuten in E. coli produzieren. Die Immunglobulindomänen der leichten Kette sind orange, die der schweren Kette rosa gefärbt. Die 6 hypervariablen Peptidschleifen, die auf beiden Seiten des intakten Antikörpers jeweils eine identische Bindungsstelle für das Antigen bilden, sind blau dargestellt. Mitte: Räumliche Überlagerung von sieben verschiedenen menschlichen Vertretern der Lipocalin-Familie (RBP, ApoD, AGP, ? 1m, NGAL, c8 ?, Tlc). Deren ?-Fass-Struktur ist hochkonserviert, wohingegen die jeweils vier Schleifen, die den Eingang zu der kelchartigen Liganden-Bindungsstelle am oberen Ende bilden, hohe Variabilität aufweisen. Ihre gerichtete strukturelle Veränderung zur molekularen Erkennung vorgegebener Zielmoleküle bildet die Grundlage der Anticalin-Technologie. Rechts: Simulation eines PASylierten Fab-Fragments. Das pharmakologisch aktive Protein ist magenta/blau – mit seiner Antigen-Bindungsstelle gelb – wiedergegeben. Die zufallsknäuelartigen Polypeptidstrukturen an dessen unterem Ende stellen insgesamt 24 verschiedene Konformationen dar, in verschiedenen Grautönen überlagert, was das aufgeblähte biophysikalische Verhalten des PAS-Anhängsels illustriert.
Der Durchbruch in der Antikörper-Technologie Nach gründlichem Studium des Kapitels über das multicistronische (also für mehrere Polypeptidketten codierende) lac-Operon in Prof. Rolf Knipper’s Lehrbuch „Molekulare Genetik“ machte ich mich anhand der gesammelten Informationen an die gentechnische Konstruktion eines künstlichen Operons für die beiden Ketten eines Antikörperfragments (zunächst Fab, später Fv), jeweils ausgestattet mit einer bakteriellen Signalsequenz. Mit einem der resultierenden Expressionsplasmide gelang mir schließlich im November 1987 der Durchbruch: Zum ersten Mal konnte ich ein bindungsaktives Fv-Fragment eines Antikörpers aus dem periplasmatischen Zellextrakt von E. coli isolieren [4]. Kritisch dafür war die streng kontrollierte Genexpression mittels Katabolitrepression durch Glucose, um toxische Effekte des Fremdproteins auf die Bakterienzelle zu unterdrücken; ein Problem, das ich erst Jahre später durch Nutzung des tet-Promotors nachhaltig lösen konnte [5]. Ein weiterer Erfolgsfaktor war die Optimierung eines affinitätschromatographischen Reinigungsschritts, wobei mich Kenntnisse aus dem früheren Studium der technischen Chemie auf die richtige Lösung brachten – selbst in dem nun viel kleineren Labormaßstab. Innerhalb weniger Wochen stellte ich alle erforderlichen Kontrollexperimente fertig. Noch vor Weihnachten desselben Jahres reichte ich zusammen mit A. Plückthun sowohl ein Manuskript bei „Science“ [4] als auch eine Patentanmeldung beim Deutschen Patentamt ein. Wie wir später erfuhren, kam eine Arbeitsgruppe bei der US-Firma INGENE (heute Xoma) nahezu gleichzeitig auf eine ähnliche technische Lösung, und beide Artikel wurden im Mai 1988 direkt hintereinander publiziert. Diese Entwicklung war der entscheidende Katalysator für das sich daraufhin rasant entfaltende Forschungsgebiet des Antikörper-Engineerings, denn damit wurde es möglich, schnell und auf einfache Weise funktionelle Antikörperfragmente in einem verbreitet eingesetzten Laborbakterium zu produzieren und durch Mutagenese gezielt zu verändern. Demgegenüber steckten Verfahren zur gentechnischen Herstellung von Antikörpern in Säugerzellkulturen seinerzeit noch in den Anfängen. Screening-Verfahren und Gensynthese für ein künstliches Antikörperfragment Nach der Promotion an der LMU Ende 1989 ging ich mit einem Stipendium im „Sonderprogramm Gentechnologie“ des DAAD an das MRC Laboratory of Molecular Biology in Cambridge, England in die Abteilung von Dr. Cesar Milstein zu Dr. Greg Winter, in dessen Labor kurz zuvor die Methode der Antikörperhumanisierung mittels „CDR-Grafting“ entwickelt worden war. Nach meiner Ankunft schlug G. Winter mir vor, die Idee für das „Phage Display“ von funktionellen Proteinen, die er von Dr. Jim Wells bei der Firma Genentech aufgeschnappt hatte, auf Antikörperfragmente zu übertragen; allerdings zog ich es vor, mich mit dem rationalen Design von Antikörpern zu beschäftigen. Dazu erlernte ich in Cambridge die noch junge Methode der Polymerasekettenreaktion, und mit dem Trick des „PCR-Assembly“ realisierte ich erstmals die Gensynthese für ein durch Computer-Modelling konstruiertes künstliches Antikörperfragment [6]. Zudem entwickelte ich ein neuartiges Screening-Verfahren für Antikörperfragmente auf der Ebene von Bakterienkolonien [7]. Prägend während meiner Zeit in Cambridge waren nicht nur die zahlreichen Gespräche mit hochrangigen Wissenschaftlern während der gemeinsamen „Coffee & Tea Breaks“ in der Institutskantine, sondern auch die intensiven Diskussionen mit meinem Mentor G. Winter, der sich derzeit mit der Ausgründung des Start-up-Unternehmens Cambridge Antibody Technologie (CAT; heute Medimmune) beschäftigte, über die kommerzielle Verwertung biotechnologischer Forschungsergebnisse. Dank eines Angebots von Prof. Hartmut Michel konnte ich im darauffolgenden Jahr am Max-Planck-Institut für Biophysik in Frankfurt/M. meine eigene Arbeitsgruppe aufbauen. In dem gut ausgestatteten Umfeld und mit Unterstützung meiner ersten Doktoranden nahm ich neue Forschungsprojekte in Angriff. Neben Anwendungen rekombinanter Antikörperfragmente zur Erforschung von Membranproteinen [8] stand dabei die Entwicklung einer neuen Proteinreinigungsmethode sowie einer Nachfolgetechnologie für Antikörper im Vordergrund. Bis Anfang der 90er-Jahre wurden in E. coli produzierte Antikörperfragmente entweder mit mehrstufigen konventionellen Chromatographieverfahren gereinigt oder unter Ausnutzung ihrer natürlichen Bindungsfunktion mittels Antigen- oder Hapten-Affinitätschromatographie. Es bestand daher dringender Bedarf an einem von den individuellen Antikörpereigenschaften unabhängigen und einfach durchführbaren Reinigungsverfahren. Zwar war es mir mit A. Plückthun zuvor gelungen, das sogenannte His-Tag in Verbindung mit der Metallchelat-Affinitätschromatographie (was bis dahin für die Reinigung rekombinanter Proteine nur unter denaturierenden Bedingungen propagiert worden war) auch auf in E. coli produzierte funktionelle Antikörperfragmente anzuwenden [9]; jedoch erwies sich die für dieses Verfahren benötigte hohe Ionenstärke als nachteilig. In Ermangelung geeigneter Antikörper war das His-Tag damals auch nicht für den gewünschten Nachweis der gentechnisch hergestellten Proteine geeignet. Ein innovatives Affinitätsanhängsel und die Suche nach Antikörperalternativen Inspiriert durch eine Publikation über Peptid-Zufallsbibliotheken entwickelte ich daher zusammen mit meinem ersten Doktoranden, Thomas Schmidt, eine Sequenz aus neun Aminosäuren, die intrinsische Bindungsaktivität für das zu diesem Zeitpunkt bereits – wegen seiner Biotin-Bindungsaktivität – weitverbreiteten Proteinreagenz Streptavidin aufwies [10]. Dieses von uns als Strep-tag® bezeichnete Affinitätsanhängsel gestattete nicht nur die überraschend effiziente Reinigung rekombinanter Proteine unter besonders schonenden Bedingungen im Einschrittverfahren, sondern es erlaubte zudem deren Nachweis im ELISA oder auf dem Western-Blot mithilfe kommerziell verfügbarer Reagenzien [11]. Diese Technologie wurde von der Max-Planck-Verwertungsgesellschaft „Garching Innovation“ zum Patent angemeldet und später an die IBA GmbH, Göttingen auslizenziert (zusammen mit dem oben erwähnten tet-Promotor). Unterstützt durch eine Reihe von Weiterentwicklungen in den darauf folgenden Jahren ist das Strep-tag heute zu einem weltweit angewandten Research Tool geworden [12]. Anlass für die Suche nach einer Alternative zur Antikörpertechnologie lieferte die Erkenntnis aus meinen vorangegangenen Arbeiten, wonach die Zusammensetzung aus zwei verschiedenen Immunglobulinketten eine grundsätzliche Erschwernis vor allem für die bakterielle Produktion und das Protein-Engineering darstellt. Die Idee für das Design gänzlich neuartiger Bindeproteine ging zurück auf eine Begegnung mit Prof. Robert Huber und Dr. Alwyn Jones noch während meiner Zeit am MPI für Biochemie, wo beide die von ihnen jeweils aufgeklärten Kristallstrukturen des Retinol-Bindungsproteins und des Bilin-Bindungsproteins miteinander verglichen. Dabei machten sie die überraschende Beobachtung, dass die beiden Proteine trotz ihrer verschiedenen Liganden-Bindungsfunktion, ihrer unterschiedlichen Herkunft (Mensch bzw. Insekt) und ihrer extrem niedrigen Aminosäure-Sequenzverwandtschaft den gleichen ß-Fass-Faltungstyp aufwiesen und sich bloß in ihrer aus vier Peptidschleifen bestehenden Liganden-Bindungsstelle strukturell unterschieden [13]. Die grundlegende Analogie dieses Proteinfaltungsprinzips mit der Kombination aus konservierter Gerüststruktur und hypervariabler Schleifenregion, die für die Antigen-Spezifität von Antikörpern verantwortlich ist, lag auf der Hand. Dagegen waren diese Vertreter der sogenannten Lipocalinproteinfamilie mit jeweils ca. 180 Aminosäuren wesentlich kleiner und sie bestanden nur aus einer einzigen Polypeptidkette. PCR-basierte Methoden für die Synthese nicht bloß von einzelnen Genen sondern sogar von gezielt randomisierten Gen-Bibliotheken waren inzwischen etabliert, und Protein-Selektionsverfahren durch Phage-Display waren populär geworden; vor diesem Hintergrund drängte sich geradezu die Vorstellung auf, die Bindungsstelle eines natürlichen Lipocalins durch gezielte Zufallsmutagenese zu variieren und aus der resultierenden „Proteothek“ künstliche Lipocalinvarianten mit neuartigen Liganden-Spezifitäten zu selektieren. Die Anticalin-Story Nach mehrjähriger Pionierarbeit unter unerwartet mühsamer Optimierung zahlreicher Einzelmethoden gelang der Durchbruch in diesem Projekt mit der Generierung eines künstlichen Lipocalins mit ausgeprägter Spezifität und hoher Affinität für den vorgegebenen Liganden Fluorescein. Die erfolgreiche Selektion und funktionelle Charakterisierung dieses ersten sogenannten Anticalins® fand an der TU Darmstadt statt, wo ich 1994 eine Professur für Proteinchemie angetreten hatte. Gleich nach der Komplettierung der experimentellen Datensätze meldeten wir diese Erfindung zum Patent an und bereiteten anschließend eine wissenschaftliche Publikation vor [14]. Nach meinem Ruf auf den Lehrstuhl für Biologische Chemie an der Technischen Universität München im Jahr 1998 war ich mit meiner wachsenden Arbeitsgruppe in der Lage, die Anticalintechnologie auszubauen und für praktische Anwendungen nutzbar zu machen [15]. Das nächste Anticalin, das wir entwickelten, hatte Spezifität für ein Digitalis-Pflanzensteroid, welches nicht nur als kardiovaskulärer Wirkstoff von medizinischem Nutzen ist sondern auch als biochemische Reportergruppe in der Forschung dient. Wesentlich für die systematische Weiterentwicklung der Anticalintechnologie war die Möglichkeit, an der TU München ein eigenes Labor für Röntgenstrukturanalyse von Proteinen aufzubauen. Damit gelang es uns, die Kristallstrukturen der ersten Anticaline aufzuklären und den Einfluss der Aminosäuremutationen auf die Gestalt und Funktion der abgewandelten Bindungsstellen in der Lipocalingerüststruktur zu analysieren. Überlegungen zur Kommerzialisierung der Anticalintechnologie begannen mit meiner Teilnahme am Münchener Businessplan-Wettbewerb (MBPW) 1999/2000, auf den ich durch eine Informationsveranstaltung an unserem Campus Weihenstephan aufmerksam geworden war. Ausgangspunkt war ein Geschäftskonzept für die biopharmazeutische Entwicklung der Anticaline, ähnlich, wie es die Biotech-Ausgründungen CAT in Cambridge sowie Morphosys in Martinsried – an deren Entstehung A. Plückthun beteiligt war – im Bereich der Antikörper-Technologie erfolgreich vorgemacht hatten. Im Verlauf des sich über neun Monate hinstreckenden dreiphasigen Wettbewerbs gestaltete sich dieses Konzept, motiviert durch Prämierungen schon während der beiden ersten Phasen, bis zum ausformulierten Businessplan. Gleichzeitig wuchs unser Team schrittweise durch Rekrutierung eines Kaufmanns, Claus Schalper, eines in der Biotech-Branche erfahrenen ehemaligen Studienkollegen, Karsten Schürrle, sowie eines in der Anticalintechnologie versierten Doktoranden, Steffen Schlehuber. Zu unserer großen Freude errangen wir mit unserem Konzept für das Biotech-Unternehmen „Pieris“ beim MBPW im Juli 2000 schließlich den ersten Preis. Die Monate danach standen unter dem Eindruck zahlreicher Treffen mit Kapitalgebern und Investoren, denen wir unsere Gründungsidee vorstellen durften und mit denen wir Finanzierungsmöglichkeiten diskutierten. Im Januar 2001 schließlich vollzogen wir die Ausgründung der „kleinen AG“ mit Beteiligung der BioM AG und der TransConnect GmbH als Seed-Investoren. Kurz danach konnten wir Dr. Martin Pöhlchen als Geschäftsführer mit Branchenerfahrung zur Verstärkung unseres Teams gewinnen. Ich selbst war dem jungen Unternehmen als Vorsitzender des Aufsichtsrats, wissenschaftlicher Berater sowie offizieller Kooperationspartner mit meinem Lehrstuhl an der TUM verbunden. Die ersten Jahre der Unternehmensentwicklung waren geprägt von dem Aufbau der Firmenorganisation am Innovations- und Gründerzentrum Biotechnologie (IZB) in Weihenstephan sowie der Sicherung weiteren Kapitals durch eine „Serie A“-Finanzierung unter Beteiligung internationaler Investoren. Daneben hatten wir uns zwei grundlegende technologische Weiterentwicklungen vorgenommen: erstens die Selektion von Anticalinen gegen Proteinantigene (im Gegensatz zu den zunächst adressierten niedermolekularen Haptenen), welche den Hauptteil der medizinisch relevanten Zielstrukturen ausmachen, und zweitens die Nutzbarmachung menschlicher Vertreter der Lipocaline als weitere Gerüststrukturen für die Selektion von Anticalinen. Gegenüber dem ursprünglich verwendeten Insekten-Lipocalin sollte sich damit eine potenzielle Immunreaktion auf entsprechende Anticalinwirkstoffe bei der Anwendung am menschlichen Patienten minimieren lassen. Dank der florierenden Grundlagenforschung an meinem Lehrstuhl und der systematischen Strukturaufklärung der bislang noch nicht analysierten natürlichen menschlichen Lipocaline konnten wir beide Problemstellungen mit nachhaltigem Erfolg lösen [16,17]. Inzwischen verfügen wir über Anticalinwirkstoffkandidaten gegen eine Reihe hochaktueller therapeutischer Targets wie z.B. VEGF-A, CTLA-4, PSMA oder das Alzheimer-Amyloid-Peptid. Pieris hat mittlerweile zwei Anticaline bis zur klinischen Prüfung gebracht; derzeit sieht ein zweites Anticalin, das gegen das an Anämie-Erkrankungen ursächlich beteiligte Hepcidin-Peptid gerichtet ist, dem Abschluss einer „Phase I-Studie“ entgegen. Auch die Firmenstruktur unserer Biotech-Ausgründung hat sich in den letzten Jahren weiterentwickelt; so erlangte das Unternehmen nach weiteren Finanzierungsrunden schließlich Zugang zum US-Kapitalmarkt, wo es nun unter dem Namen Pieris Pharmaceuticals, Inc. an der Nasdaq gelistet ist. Proteine in „XL“ Eine grundsätzliche Problematik, die bei der Entwicklung von Anticalinen für die klinische Anwendung zutage trat, betraf deren sehr kurze Zirkulationsdauer im Körper. Generell unterliegen alle kleinen bis mittelgroßen Proteine einer effizienten Filtration in der Niere, was zur schnellen Ausscheidung aus dem Blutstrom führt. Eine Ausnahme davon bilden intakte Antikörper, die nicht nur aufgrund ihrer enormen Molekülgröße schlecht filtrierbar sind, sondern die zudem einem speziellen Rückhaltemechanismus (endosomales Recycling) unterliegen. Um die kurze Plasmahalbwertszeit von Hormonen, Cytokinen und anderen protein- oder peptidbasierten Wirkstoffen zu verlängern, wurde in den 90er-Jahren die chemische Kopplung mit dem synthetischen Polyethylenglycol (PEG) entwickelt. Diese Methodik war auch schon Thema einer Fachtagung über „Recombinant Antibodies“ in Berlin, auf der ich Anfang Juni 2005 einen Vortrag über die Anticalintechnologie hielt. Dabei herrschte der Tenor, dass die PEGylierung kostspielig, technisch aufwendig und mit Verlusten an Ausbeute und Aktivität für den biotechnologischen Wirkstoff verbunden ist. Vor diesem Hintergrund erschien mir die Idee, ob man als Alternative für ein hoch wasserlösliches, strukturell ungeordnetes Polymer wie PEG nicht auch eine einfache und chemisch durchaus ähnlich aufgebaute Aminosäuresequenz verwenden könnte, beispielsweise Polyglycin. Solche Aminosäuren ließen sich genetisch codieren und so direkt im Verbund mit dem eigentlichen Proteinwirkstoff gentechnisch herstellen. Zwar konnte dieses Konzept in meinem Labor relativ schnell umgesetzt werden [18], jedoch zeigte das biosynthetische Polyglycin nur begrenzten Erfolg, da mit zunehmender Länge des Polypeptids die Löslichkeit des Fusionsproteins erheblich abnahm. Vom Prinzip überzeugt, untersuchten wir daraufhin verwandte Aminosäuresequenzen. Tatsächlich konnten wir nach wenigen Versuchen ein Polypeptid aus Prolin, Alanin und Serin (PAS) identifizieren, das in geradezu überraschender Weise die gewünschten PEG-artigen Eigenschaften zeigte [19]. Studien mit typischen biopharmazeutischen Wirkstoffen – z.B. Wachstumshormone, Interferone, Antikörperfragmente und auch Lipocaline – ergaben, dass die PAS-Sequenzen unabhängig vom Fusionspartner eine drastische Vergrößerung der scheinbaren Molekülgröße bewirkten, was zu einer erheblich verlängerten Plasmahalbwertszeit führte. Gleichzeitig blieb die biologische Wirkung der Proteine erhalten und die PAS-Sequenzen selbst zeigten keinerlei toxische oder immunogene Wirkung im Tierversuch. Die PASylation®-Technologie wurde im Jahr 2007 von der TU München zum Patent angemeldet. Es war klar, dass diese PEG-Alternative breites Anwendungspotenzial eröffnet, so dass sich hierfür ein eigenes Geschäftsmodell anbot. Dies galt umso mehr, als inzwischen auch Daten bekannt wurden, die die Gefahr einer Anreicherung des synthetischen PEG in Organen belegten – ein Problem, das für das aus natürlichen Aminosäuren aufgebaute, biologisch abbaubare PAS-Polypeptid grundsätzlich nicht besteht. Zusammen mit meinem früheren Mitgründer C. Schalper und zwei Mitarbeitern meines Lehrstuhls, Uli Binder und Dr. Martin Schlapschy, gründete ich daraufhin die XL-protein GmbH, welche die Schutzrechte von der TUM – vermittelt durch die Bayerische Patentallianz GmbH – exklusiv einlizenzierte. Die XL-protein ist im Weihenstephaner IZB untergebracht, betreibt eigene Entwicklungsprogramme und unterhält eine wachsende Zahl von Partnerschaften mit internationalen Pharma- und Biotechunternehmen [20]. Technologie und Unternehmenskonzept wurden im Jahr 2010 jeweils mit dem 1.?Preis beim Science4Life Business-Plan Wettbewerb (Frankfurt/M.) und beim Universal Biotech „Prize of Innovation“ (Paris) prämiert. Die Vorbereitung eines ersten PASylierten Wirkstoffkandidaten auf die klinische Prüfung ist im Gange. Fazit eines unternehmerischen Professors Für mich hat sich eine solide naturwissenschaftliche Ausbildung – vor allem in der Chemie und Biochemie mit dem erlernten analytischen Denkvermögen – in Verbindung mit einem in der Praxis gewachsenen Geschäftssinn als die ideale Voraussetzung für erfolgreiche Unternehmungsgründungen auf dem Gebiet der Biotechnologie erwiesen. Ich würde mir wünschen, dass mehr junge Wissenschaftler ihr Fachwissen und Potenzial ebenfalls für die Umsetzung von Ergebnissen aus der Grundlagenforschung in die wirtschaftliche Anwendung nutzen.
Literatur |
L&M 8 / 2015
Das komplette Heft zum kostenlosen Download finden Sie hier: zum Download Der Autor:Weitere Artikel online lesen



NewsSchnell und einfach die passende Trennsäule finden
© Text und Bild: Altmann Analytik ZEISS stellt neue Stereomikroskope vor
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen. © Text und Bild: Carl Zeiss Microscopy GmbH |





