L&M-2-2009
>
Chiralität - Biologische Homochiralität und asymmetrische Synthese
Chiralität - Biologische Homochiralität und asymmetrische Synthese
Prof. Dr. Michael Reggelin,
Clemens-Schöpf-Institut für Organische Chemie und Biochemie,
Technische Universität Darmstadt
Die Chemie handelt von der Manipulation dreidimensionaler Objekte. Deshalb wird jede chemische Transformation durch die räumliche Anordnung der Atome in Molekülen beeinflusst, was wiederum bedeutet, dass Chemie immer Stereochemie ist! Wenn wir darüber hinaus diese räumliche Anordnung als den Schlüssel zur Beschreibung von Materie auf atomarer Ebene ansehen (was eine unzulässige Vereinfachung ist!), dann ist diese die Basis des Konzeptes „Struktur“ mit dem wir Chemiker unsere Moleküle charakterisieren. Wie dürfen auch davon ausgehen, dass in der Struktur der Moleküle und in höher organisierten Zuständen der Materie deren Funktion begründet liegt. Struktur kodiert für Funktion und umgekehrt wird Funktion durch Struktur realisiert. Es ist das Privileg des Chemikers Strukturen manipulieren zu können. Durch Synthese baut er neue Strukturen auf und Strukturen motivieren ihn zur Synthese. Damit ist er prinzipiell in der Lage gezielt Funktionen von Wirk- und Werkstoffen durch Synthese zu realisieren.

Chiralen Objekten, d.h. Objekten, die sich durch die Abwesenheit von n-zähligen Drehspiegelachsen auszeichnen, kommt hierbei eine besondere Bedeutung zu. Seit den Tagen L. Pasteurs erkannte man in zunehmendem Maße einen fundamentalen Zusammenhang zwischen chiralen Molekülen und lebender Materie. Mehr noch, fast alle biologisch relevanten Moleküle und darunter insbesondere die Biopolymere sind nicht nur chiral, sondern auch einheitlich konfiguriert, weisen also alle eine bestimmte Händigkeit auf. Diese sogenannte „biologische Homochiralität“ wie sie etwa durch die exklusive Verwendung von L-Aminosäuren und D-Zuckern in Proteinen bzw. Nucleinsäuren zum Ausdruck kommt, ist nicht nur fundamental mit der Entstehung des Lebens auf unserem Planeten verknüpft, sondern wirft darüber hinaus Fragen auf, die bis auf die Ebene der Elementarteilchenphysik reichen.
Paritätsverletzung und chirale Moleküle
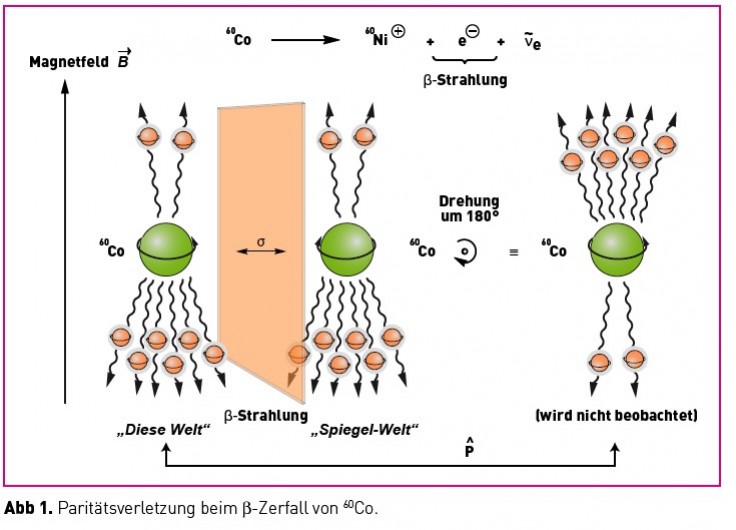
Die Beschreibung der räumlichen Symmetrie von Objekten kann durch Analyse ihrer Symmetrieelemente erfolgen. Chirale Objekte zeichnen sich dabei durch die Abwesenheit von Spiegelsymmetrien (Sn) aus, was gleichzeitig bedeutet, dass Chiralität sehr wohl mit der Anwesenheit n-zähliger Drehachsen (Cn) kompatibel ist. Von fundamentalerer Bedeutung für die Beschreibung chiraler Objekte sind jedoch nicht diese räumlichen Symmetrien, sondern die Symmetrien in den physikalischen Gesetzen, die diese Objekte beschreiben. Dabei spielen die Parität (P), die Zeitumkehr (T) und die Ladungsumkehr (C) eine besondere Rolle. Die Paritätsoperation (P) invertiert die Koordinaten eines Systems durch den Koordinatenursprung (r →-r). Diese Operation ist identisch mit der Spiegelung des Systems an einer beliebigen Ebene, gefolgt von einer Rotation um 180 ° um eine Achse senkrecht zu dieser Ebene. Zeitumkehr (T) invertiert die Bewegungsrichtung aller Teilchen eines Systems und Ladungsumkehr (C) überführt Teilchen in Antiteilchen. Bis 1956 bestand Konsens darüber, dass alle physikalischen Gesetze invariant bezüglich P, T und C sind. Daraus folgt, dass es keine bevorzugte Händigkeit chiraler Objekte in der Natur geben sollte. Diese Vorstellung fand ihr jähes Ende durch ein Experiment, mit dem die Richtungsverteilung der beim radioaktiven Zerfall (â-Zerfall) von 60Co emittierten Elektronen ermittelt wurde. Gemäß einer Vorhersage von Lee und Yang aus eben diesem Jahre sollte die für den â-Zerfall verantwortliche „schwache Kraft" eine paritätsverletzende Wechselwirkung sein. [1] Nur ein Jahr später zeigten Wu et al., dass in einem starken Magnetfeld bei tiefer Temperatur (0.1 K) ausgerichtete, â-strahlende 60Co-Kerne Elektronen bevorzugt mit einer Impulskomponente antiparallel zum Magnetfeld emittieren (Abb. 1) [2].
 Da dieses Verhalten nicht invariant unter Spiegelung ist, war die Paritätsverletzung der schwachen Wechselwirkung gezeigt. Oder: Der â-Zerfall führt zu longitudinal polarisierten Elektronen, die man als „linkshändig“ bezeichnen kann, da ihre Spins antiparallel zur Ausbreitungsrichtung orientiert sind. Diese Tatsache führt zu einer Reihe weit reichender Konsequenzen. Zunächst einmal ergibt sich daraus so etwas wie eine universelle optische Aktivität (Händigkeit) als Folge des durch die Paritätsverletzung hervorgerufenen inhärent bevorzugten Chiralitätssinnes der Atome! Mehr noch: Als Konsequenz der in den 60 er-Jahren entwickelten Theorie der elektroschwachen Wechselwirkung elektroschwache WW (einer Vereinigung der elektromagnetischen mit der schwachen Wechselwirkung) [3] kommt es zu einer Infiltration der elektromagnetischen Phänomene durch die Händigkeit der schwachen Kraft, wodurch letztere in die Welt der Moleküle und damit in diejenige der Chemie eindringt. Eine dramatische Folge obiger Diskussion ist etwa die Aufhebung der energetischen Entartung von Spiegelbildisomeren! Ein Enantiomer aus Teilchen ist im präzisen Sinne des Wortes ist unter Berücksichtigung der paritätsverletzenden schwachen Wechselwirkung ein Spiegelbildisomer aus Antiteilchen (die Kombination aus C und P ist erhalten). Die Energiedifferenz („parity violating energy difference“, PVED) zwischen Spiegelbildisomeren aus Materie ist sehr klein (10-17 bis 10-14 kT). Der sich daraus berechnende Enantiomerenüberschuss (ee) ergibt 106 bis 109 Moleküle eines „Enantiomeren“ pro Mol Racemat (10-16 – 10-13 % ee!).
Da dieses Verhalten nicht invariant unter Spiegelung ist, war die Paritätsverletzung der schwachen Wechselwirkung gezeigt. Oder: Der â-Zerfall führt zu longitudinal polarisierten Elektronen, die man als „linkshändig“ bezeichnen kann, da ihre Spins antiparallel zur Ausbreitungsrichtung orientiert sind. Diese Tatsache führt zu einer Reihe weit reichender Konsequenzen. Zunächst einmal ergibt sich daraus so etwas wie eine universelle optische Aktivität (Händigkeit) als Folge des durch die Paritätsverletzung hervorgerufenen inhärent bevorzugten Chiralitätssinnes der Atome! Mehr noch: Als Konsequenz der in den 60 er-Jahren entwickelten Theorie der elektroschwachen Wechselwirkung elektroschwache WW (einer Vereinigung der elektromagnetischen mit der schwachen Wechselwirkung) [3] kommt es zu einer Infiltration der elektromagnetischen Phänomene durch die Händigkeit der schwachen Kraft, wodurch letztere in die Welt der Moleküle und damit in diejenige der Chemie eindringt. Eine dramatische Folge obiger Diskussion ist etwa die Aufhebung der energetischen Entartung von Spiegelbildisomeren! Ein Enantiomer aus Teilchen ist im präzisen Sinne des Wortes ist unter Berücksichtigung der paritätsverletzenden schwachen Wechselwirkung ein Spiegelbildisomer aus Antiteilchen (die Kombination aus C und P ist erhalten). Die Energiedifferenz („parity violating energy difference“, PVED) zwischen Spiegelbildisomeren aus Materie ist sehr klein (10-17 bis 10-14 kT). Der sich daraus berechnende Enantiomerenüberschuss (ee) ergibt 106 bis 109 Moleküle eines „Enantiomeren“ pro Mol Racemat (10-16 – 10-13 % ee!).
Dennoch hat dies schon sehr früh zu Spekulationen geführt, die diese winzige Unausgeglichenheit mit dem Ursprung der biologischen Homochiralität in Verbindung bringt. Bereits 1959 schlugen Vester und Ulbricht einen Mechanismus vor, der, ausgehend von der longitudinalen Polarisation der β-Strahlung, zu einer Enantiomeren- differenzierenden Radiolyse von Racematen via circular polarisierter Bremsstrahlung führen sollte [4,5]. Trotz langjähriger Experimente konnte diese Hypothese jedoch bis heute nicht zweifelsfrei verifiziert werden [6]. *Absolute asymmetrische Synthese, initialer Symmetriebruch* Aus der bevorzugten Richtung mit der die Elektronen beim β-Zerfall emittiert werden, kann man ableiten, dass das Skalarprodukt aus s (Spin) und p (Impuls) einen von Null verschiedenen Wert annimmt (eine Helizität h). Da dieses Produkt aus einem polaren (p) und einem axialen Vektor (s) ein Zeit-gerader (time-even) Pseudoskalar (ein vorzeichenbehafteter Skalar, Spatprodukt) ist, kann man dessen Auftreten als Charakteristikum von Chiralität definieren („wahre Chiralität“ – true chirality) [7, 8]. Oder: „Wahre Chiralität tritt in Systemen auf, die in durch Rauminversion (P) interkonvertierbaren Zuständen existieren können, aber nicht durch eine Kombination aus Zeitumkehr (T) und Rotation (R) auseinander hervorgehen dürfen. P darf nicht durch T rückgängig gemacht werden können (Abb. 2).
 Dieser zweite Teil der Definition von „wahrer Chiralität“ ist irrelevant für stationäre Objekte, hat aber wichtige Implikationen für die Suche nach „wahrhaft chiralen Einflüssen“, also solchen, die die Symmetrie eines Racemates oder einer achiralen Verbindung brechen können. Dies trifft weder für elektrische (E) noch magnetische Felder (B) sowie irgendeine Kombination der beiden zu, da dann zwar eine polare/axiale Kombination vorliegt, E und B aber unterschiedlich unter T transformieren (eine Punktspiegelung kann also durch Zeitumkehr rückgängig gemacht werden („falsche Chiralität“– „false chirality“). Ein „wahrhaft chiraler Einfluss" tritt jedoch auf, wenn ein Lichtstrahl mit Propagationsvektor k (polar, Zeit-ungerade) parallel oder antiparallel zu einem magnetischen Feld (B, axial, Zeit-ungerade) kombiniert wird. Unter dem Einfluss dieser Kombination erwartet man beim Durchstrahlen einer Lösung von einheitlich konfigurierten chiralen Molekülen eine Abhängigkeit der Absorptionskoeffizienten (∑) von der relativen Anordnung zwischen B und k. In der Tat wurde diese „magnetochirale Anisotropie“ [∑ (↑ ↑) – ∑ ( ↑ ↓) ≠ 0] im Jahre 2000 durch Rikken und Raupach experimentell verifiziert [9].
Dieser zweite Teil der Definition von „wahrer Chiralität“ ist irrelevant für stationäre Objekte, hat aber wichtige Implikationen für die Suche nach „wahrhaft chiralen Einflüssen“, also solchen, die die Symmetrie eines Racemates oder einer achiralen Verbindung brechen können. Dies trifft weder für elektrische (E) noch magnetische Felder (B) sowie irgendeine Kombination der beiden zu, da dann zwar eine polare/axiale Kombination vorliegt, E und B aber unterschiedlich unter T transformieren (eine Punktspiegelung kann also durch Zeitumkehr rückgängig gemacht werden („falsche Chiralität“– „false chirality“). Ein „wahrhaft chiraler Einfluss" tritt jedoch auf, wenn ein Lichtstrahl mit Propagationsvektor k (polar, Zeit-ungerade) parallel oder antiparallel zu einem magnetischen Feld (B, axial, Zeit-ungerade) kombiniert wird. Unter dem Einfluss dieser Kombination erwartet man beim Durchstrahlen einer Lösung von einheitlich konfigurierten chiralen Molekülen eine Abhängigkeit der Absorptionskoeffizienten (∑) von der relativen Anordnung zwischen B und k. In der Tat wurde diese „magnetochirale Anisotropie“ [∑ (↑ ↑) – ∑ ( ↑ ↓) ≠ 0] im Jahre 2000 durch Rikken und Raupach experimentell verifiziert [9].
Neben der elektroschwachen Wechselwirkung existiert also mit diesem magnetochiralen Effekt eine weitere fundamentale Ursache für den initialen Symmetriebruch, der letztlich zur biologischen Homochiralität geführt haben könnte. Freilich sind diese Effekte sehr klein und es sind beileibe nicht die einzigen, die diskutiert werden (inklusiver solcher extraterrestrischer Natur). Wie dem auch immer gewesen sei, nach dem initialen Symmetriebruch gab es vermutlich zunächst chirale Moleküle mit sehr geringem Enantiomerenüberschuss (ee). Daraus ergibt sich die Konsequenz, dass es Mechanismen zur Verstärkung und zum Erhalt der optischen Reinheit gegeben haben muss. Auch in diesem Bereich gibt es weit mehr Szenarien als hier diskutiert werden können. Aus Gründen, die weiter unten klar werden, sollen Polymere als mögliche Mediatoren für diese Prozesse herausgegriffen werden.
Chiralitätsverstärkung in Makromolekülen
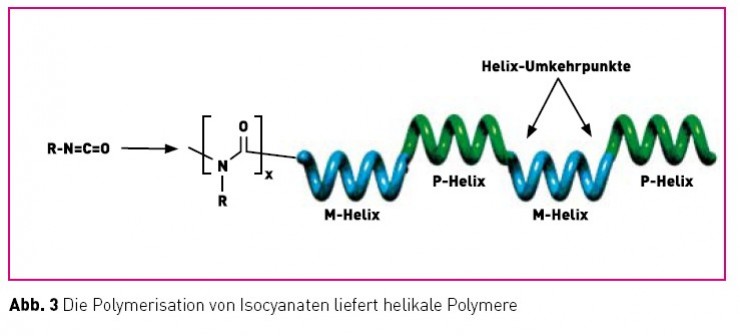
Ein für die Struktur und Funktion vonBiomakromolekülen wie Proteinen, Nukleinsäuren und Polysacchariden entscheidendes Strukturmotiv ist das einer stereoregulären Helix. Die einsinnige Händigkeit dieser Helices wird dabei hervorgerufen durch die einheitliche Konfiguration der sie konstituierenden Bausteine (Aminosäuren, Zucker). Interessanterweise gibt es aber auch eine Reihe von synthetischen Polymeren, die in helikale Konformationen falten. Für den bei Biopolymeren nicht auftretenden Fall, dass die Wiederholungseinheiten in diesen Polymeren sämtlich achiral sind ( unter Vernachlässigung der schwachen Kraft!), muss auch das resultierende Polymer optisch inaktiv sein, daher müssen die Anteile an links- bzw. rechtsgängigen Abschnitten übereinstimmen. Dies kann auf zwei unterschiedlichen Wegen realisiert werden. Entweder liegt ein äquimolares Gemisch von entgegengesetzt einhändig helikalen Ketten vor (eine Art Konglomerat) oder es bilden sich lange Ketten bestehend aus sich kompensierenden links- und rechtshelikalen Abschnitten aus, die durch mobile Helix- Umkehrpunkte voneinander getrennt sind. Für die weitere Diskussion sind nun solche Polymere interessant, für die letzteres zutrifft und die niedrige Helix-Inversionsbarrieren aufweisen (ca. 3 – 5 kcal/Mol). Diese Bedingung ist unter anderem bei Polyisocyanaten erfüllt (Abb. 3). Die Polymerisation
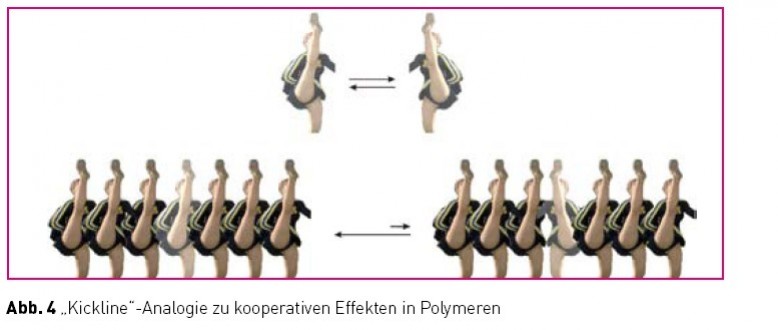
 von Isocyanaten (RNCO) liefert Polymere, die aufgrund ihrer Helix-Inversionsbarriere nur alle 600 – 800 Wiederholungseinheiten einen Umkehrpunkt aufweisen [10]. Aufgrund deren Mobilität entstehen dynamisch-helikale Objekte aus interkonvertierenden linksund rechts-helikalen Abschnitten. Durch die konformationelle Synchronisation (korrelierte Bewegungen) in den langen helikalen Segmenten können nun kleine „chirale Störungen“, etwa hervorgerufen durch die Anwesenheit geringer Mengen chiraler, nicht-racemischer Monomere, extrem verstärkt werden und somit das Polymer in einen einhändig helikalen Zustand überführen! Eine Serie von Damen, die korrelierte Bewegungen ausführen („kickline“) verhält sich analog (Abb. 4) [11].
von Isocyanaten (RNCO) liefert Polymere, die aufgrund ihrer Helix-Inversionsbarriere nur alle 600 – 800 Wiederholungseinheiten einen Umkehrpunkt aufweisen [10]. Aufgrund deren Mobilität entstehen dynamisch-helikale Objekte aus interkonvertierenden linksund rechts-helikalen Abschnitten. Durch die konformationelle Synchronisation (korrelierte Bewegungen) in den langen helikalen Segmenten können nun kleine „chirale Störungen“, etwa hervorgerufen durch die Anwesenheit geringer Mengen chiraler, nicht-racemischer Monomere, extrem verstärkt werden und somit das Polymer in einen einhändig helikalen Zustand überführen! Eine Serie von Damen, die korrelierte Bewegungen ausführen („kickline“) verhält sich analog (Abb. 4) [11].
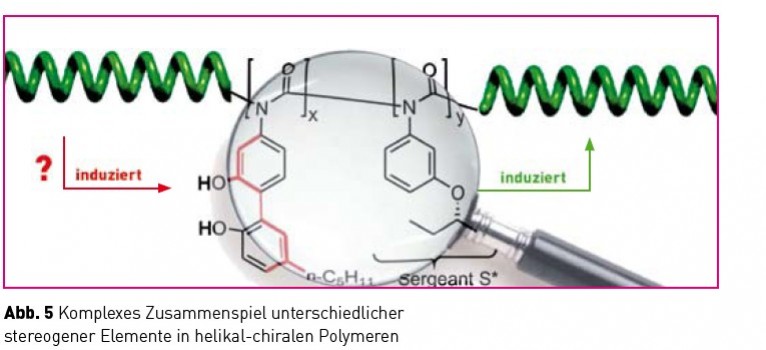
 Während die beiden allein stehenden Damen völlig ungestört sowohl ihr rechtes als auch ihr linkes Bein schwingen können, führt ihre Eingliederung in eine synchron schwingende Gruppe zu einer passenden und einer unpassenden Situation. Nach einem höflichen aber bestimmten Hinweis an die Falschtänzerin wird man sich einig und somit wird aus einer kleinen Störung (eine Tänzerin) eine Störungsfront (eine uniform schwingende Damengruppe). Diese setzt sich nun mit verstärktem Nachdruck gegenüber allen anderen Tänzerinnen durch (hier nicht gezeigt), was letztlich zu einer einheitlich schwingenden „kickline“ (einhändig helikales Polymer!) führt. Dieser Amplifikationsmechanismus wird „seargents and soldier Effekt“ [10] genannt, da eine kleine Anzahl enantiomerenangereicherter Comonomere eine große Anzahl von Wiederholungseinheiten in eine einhändige Anordnung zwingt. Ob und wenn ja welche Rolle Effekte dieser Art für die Genese der biologischen Homochiralität gespielt haben, ist eine offene Frage, ein interessanter Kandidat zu ihrer Klärung sind diese Effekte aber allemal. Wenn wir einmal davon ausgehen, dass synchronisierte konformationelle Fluktuationen unter dem Einfluss geringfügiger asymmetrischer Störungen zu einhändig helikalen Strukturen geführt haben, was passierte dann? Wie wurde diese Asymmetrie auf all die anderen heute bedeutsamen chiralen Moleküle übertragen? Nun, aufgrund der Einhändigkeit dieser Strukturen muss es prinzipiell möglich sein, diese als asymmetrischen Einfluss bei chirogenen Reaktionen einzusetzen. Diese Überlegung ist nicht nur im Zusammenhang mit der Frage der biologischen Homochiralität von Interesse, sondern darüber hinaus auch für den Entwurf neuartiger, polymerer asymmetrischer Katalysatoren für die Synthese enantiomerenreiner Verbindungen. Bemerkenswerterweise spielt dieser letztere Aspekt, trotz des enormen Forschungsaufwandes im Bereich der asymmetrischen Synthese, in der kontemporären chemischen Literatur kaum eine Rolle. Seit einiger Zeit verfolgen wir den Ansatz sowohl die bereits eingeführten dynamischen (Polyisocyanate, [12] Polyacetylene) als auch statisch-helikale Polymere (Polymethacrylate, [13] Polychinoxaline) in asymmetrischen Synthesen einzusetzen. Zu diesem Zweck haben wir die folgende Strategie entwickelt (Abb. 5).
Während die beiden allein stehenden Damen völlig ungestört sowohl ihr rechtes als auch ihr linkes Bein schwingen können, führt ihre Eingliederung in eine synchron schwingende Gruppe zu einer passenden und einer unpassenden Situation. Nach einem höflichen aber bestimmten Hinweis an die Falschtänzerin wird man sich einig und somit wird aus einer kleinen Störung (eine Tänzerin) eine Störungsfront (eine uniform schwingende Damengruppe). Diese setzt sich nun mit verstärktem Nachdruck gegenüber allen anderen Tänzerinnen durch (hier nicht gezeigt), was letztlich zu einer einheitlich schwingenden „kickline“ (einhändig helikales Polymer!) führt. Dieser Amplifikationsmechanismus wird „seargents and soldier Effekt“ [10] genannt, da eine kleine Anzahl enantiomerenangereicherter Comonomere eine große Anzahl von Wiederholungseinheiten in eine einhändige Anordnung zwingt. Ob und wenn ja welche Rolle Effekte dieser Art für die Genese der biologischen Homochiralität gespielt haben, ist eine offene Frage, ein interessanter Kandidat zu ihrer Klärung sind diese Effekte aber allemal. Wenn wir einmal davon ausgehen, dass synchronisierte konformationelle Fluktuationen unter dem Einfluss geringfügiger asymmetrischer Störungen zu einhändig helikalen Strukturen geführt haben, was passierte dann? Wie wurde diese Asymmetrie auf all die anderen heute bedeutsamen chiralen Moleküle übertragen? Nun, aufgrund der Einhändigkeit dieser Strukturen muss es prinzipiell möglich sein, diese als asymmetrischen Einfluss bei chirogenen Reaktionen einzusetzen. Diese Überlegung ist nicht nur im Zusammenhang mit der Frage der biologischen Homochiralität von Interesse, sondern darüber hinaus auch für den Entwurf neuartiger, polymerer asymmetrischer Katalysatoren für die Synthese enantiomerenreiner Verbindungen. Bemerkenswerterweise spielt dieser letztere Aspekt, trotz des enormen Forschungsaufwandes im Bereich der asymmetrischen Synthese, in der kontemporären chemischen Literatur kaum eine Rolle. Seit einiger Zeit verfolgen wir den Ansatz sowohl die bereits eingeführten dynamischen (Polyisocyanate, [12] Polyacetylene) als auch statisch-helikale Polymere (Polymethacrylate, [13] Polychinoxaline) in asymmetrischen Synthesen einzusetzen. Zu diesem Zweck haben wir die folgende Strategie entwickelt (Abb. 5).
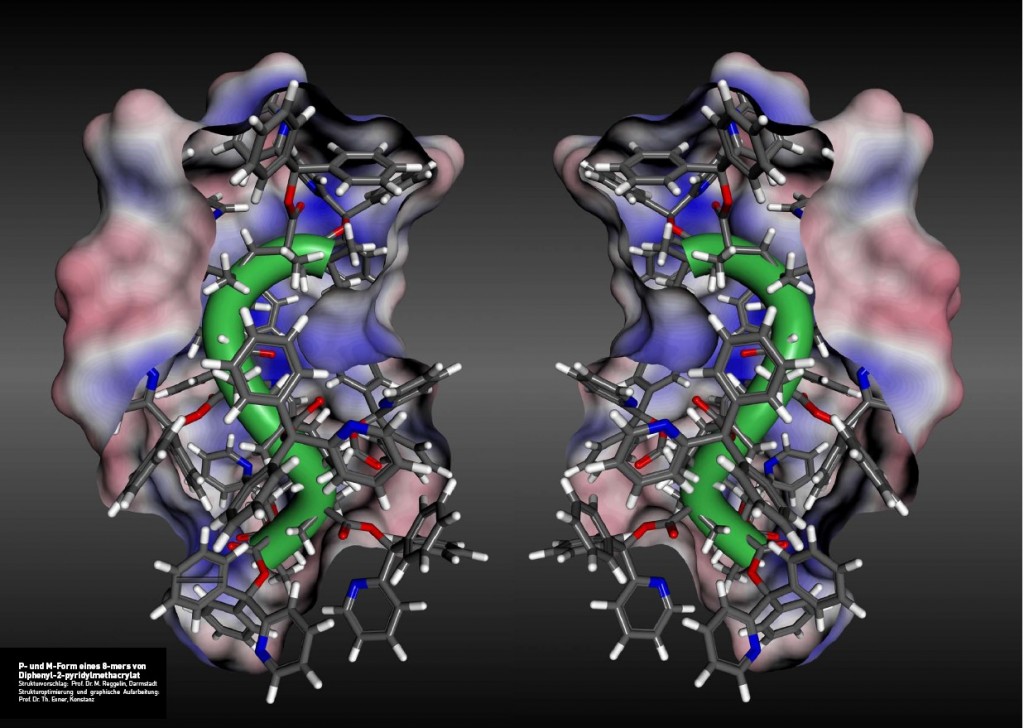
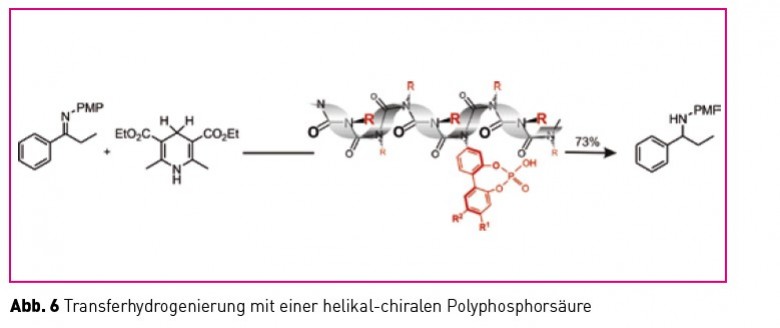
 Ein chiraler seargent induziert auf die beschriebene Weise ein einhändiges Polymer (grün), dieses wiederum beeinflusst eine torsionslabile Biarylachse (rot). Je nach induziertem sinn der Achse entstehen so diastereomorphe (energetisch unterschiedliche) Konformationen, die unterschiedlich rasch Substrate zu enantiomerenangereicherten Produkten umsetzen können sollten. In der Tat ist es uns gelungen solche Polymere herzustellen und den Nachweis ihrer katalytischen Aktivität zu führen (Abb. 6).
Ein chiraler seargent induziert auf die beschriebene Weise ein einhändiges Polymer (grün), dieses wiederum beeinflusst eine torsionslabile Biarylachse (rot). Je nach induziertem sinn der Achse entstehen so diastereomorphe (energetisch unterschiedliche) Konformationen, die unterschiedlich rasch Substrate zu enantiomerenangereicherten Produkten umsetzen können sollten. In der Tat ist es uns gelungen solche Polymere herzustellen und den Nachweis ihrer katalytischen Aktivität zu führen (Abb. 6).
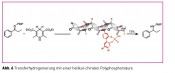 Leider kommt es im Falle der eingesetzten Polyphosphorsäure zu einer stereochemischen Komplikation (Rückkopplung der labilen Achse auf die Helix, wodurch diese ihre Uniformität verliert), die eine asymmetrische Induktion auf das neu geschaffene stereogene Zentrum im Produkt verhindert. Diese (interessante!) Unannehmlichkeit zu verstehen, um sie dann zu verhindern (evtl. mit einem anderen Polymer) ist Gegenstand aktueller Arbeiten. Die Natur nutzt Polymere seit Milliarden von Jahren zur Realisierung von Gerüststrukturen, funktionalen Strukturen (Biokatalysatoren), zur Informationsspeicherung und -weitergabe. Die Chiralität dieser Polymere spielt dabei eine entscheidende Rolle und es erscheint intuitiv plausibel und Erfolg versprechend, synthetische chirale Polymere zur Realisierung komplexer chemischer Probleme einzusetzen. Die Tatsache, dass dieser eher utilitaristische Ansatz mit fundamentalen Fragestellungen im Zusammenhang mit dem Phänomen Chiralität an sich und dem Problem der biologischen Homochiralität im Speziellen so wunderbar harmoniert, macht Forschung in diesem Spannungsfeld so ungemein auf- und anregend!
Leider kommt es im Falle der eingesetzten Polyphosphorsäure zu einer stereochemischen Komplikation (Rückkopplung der labilen Achse auf die Helix, wodurch diese ihre Uniformität verliert), die eine asymmetrische Induktion auf das neu geschaffene stereogene Zentrum im Produkt verhindert. Diese (interessante!) Unannehmlichkeit zu verstehen, um sie dann zu verhindern (evtl. mit einem anderen Polymer) ist Gegenstand aktueller Arbeiten. Die Natur nutzt Polymere seit Milliarden von Jahren zur Realisierung von Gerüststrukturen, funktionalen Strukturen (Biokatalysatoren), zur Informationsspeicherung und -weitergabe. Die Chiralität dieser Polymere spielt dabei eine entscheidende Rolle und es erscheint intuitiv plausibel und Erfolg versprechend, synthetische chirale Polymere zur Realisierung komplexer chemischer Probleme einzusetzen. Die Tatsache, dass dieser eher utilitaristische Ansatz mit fundamentalen Fragestellungen im Zusammenhang mit dem Phänomen Chiralität an sich und dem Problem der biologischen Homochiralität im Speziellen so wunderbar harmoniert, macht Forschung in diesem Spannungsfeld so ungemein auf- und anregend!
Fotos: © Prof. Dr. Michael Reggelin
Lit
[1] T. D. Lee, C. N. Yang, Physical Review 1956, 104, 254.
[2] C. S. Wu, E. Ambler, R. W. Hayward, D. D. Hoppes, R. P. Hudson, Physical Review 1957, 105, 1413.
[3] K. Gottfried, V. F. Weisskopf, Concepts of Particle Physics, Vol. 1, Oxford University Press, Oxford, England, 1984.
[4] F. Vester, T. L. V. Ulbricht, H. Krauch, Naturwissenschaften 1959, 46, 68.
[5] T. L. Ulbricht, F. Vester, Tetrahedron 1962, 18, 629.
[6] W. A. Bonner, Chirality 2000, 12, 114.
[7] L. D. Barron, Chem Soc Rev 1986, 15, 189.
[8] L. D. Barron, J Am Chem Soc 1986, 108, 5539.
[9] G. Rikken, E. Raupach, Nature 2000, 405, 932.
[10] M. M. Green, J.-W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger, J. V. Selinger, Angew. Chem. 1999, 111, 3328.
[11] J. W. Lockman, N. M. Paul, J. R. Parquette, Prog. Polym. Sci. 2005, 30, 423.
[12] M. Reggelin, S. Dörr, M. Klussmann, M. Schultz, M. Holbach, Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5461.
[13] M. Reggelin, M. Schultz, M. Holbach, Angew. Chem. Int. Ed. 2002, 41, 1614
|
L&M 2 / 2009
Diese Artikel wurden veröffentlicht in Ausgabe L&M 2 / 2009.
Das komplette Heft zum kostenlosen Download finden Sie hier:
zum Download
Der Autor:
Weitere Artikel online lesen
News
Mit dem HPLC-Säulenkonfigurator unter www.analytics-shop.com können Sie stets die passende Säule für jedes Trennproblem finden. Dank innovativer Filtermöglichkeiten können Sie in Sekundenschnelle nach gewünschtem Durchmesser, Länge, Porengröße, Säulenbezeichnung u.v.m. selektieren. So erhalten Sie aus über 70.000 verschiedenen HPLC-Säulen das passende Ergebnis für Ihre Anwendung und können zwischen allen gängigen Herstellern wie Agilent, Waters, ThermoScientific, Merck, Sigma-Aldrich, Chiral, Macherey-Nagel u.v.a. wählen. Ergänzend stehen Ihnen die HPLC-Experten von Altmann Analytik beratend zur Seite – testen Sie jetzt den kostenlosen HPLC-Säulenkonfigurator!
© Text und Bild: Altmann Analytik
Aufnahme, Dokumentation und Teilen von Ergebnissen mit ZEISS Stemi 305 und ZEISS Stemi 508
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen.
© Text und Bild: Carl Zeiss Microscopy GmbH
|