


|
Quantenchemische Berechnung von NMR-chemischen Verschiebungen in Proteinen: ein hilfreiches Tool für Experiment und Theorie?
Quantenchemische Berechnung von NMR-chemischen Verschiebungen in Proteinen: ein hilfreiches Tool für Experiment und Theorie?Reise ins UnerwarteteAnnähernd alle chemischen, biochemischen und pharmazeutischen Projekte werden heutzutage von theoretischen Berechnungen und Computersimulationen begleitet. Meistens werden diese Rechnungen und die entsprechenden Experimente aber in unterschiedlichen Gruppen durchgeführt und die Interaktion der Gruppen beschränkt sich auf den Vergleich der Ergebnisse. Hier soll am Beispiel der quantenchemischen Berechnung von NMR-chemischen Verschiebungen gezeigt werden, dass die intensive Integration der Expertise aller Seiten nicht nur zur besseren Übereinstimmung der Ergebnisse und zu neuen Erkenntnissen über das Experiment führen kann, sondern auch Möglichkeiten eröffnet, die zu vollkommen unerwarteten Anwendungsgebieten in der theoretischen Chemie führen. Die Datenbasis 
Die Strukturbestimmung von Proteinen, DNA und RNA und ihrer Komplexe untereinander sowie mit kleinen organischen Molekülen ermöglichte und ermöglicht eine Explosion der Erkenntnisse zu biochemischen Prozessen. Ein ausgezeichnetes Beispiel ist die von Watson und Crick aufgeklärte Doppelhelix-Struktur von DNA, womit die Speicher- und Kopiervorgänge des Erbguts erklärt werden konnten. Zwei experimentelle Methoden werden heutzutage für die Strukturbestimmung in atomarer Auflösung herangezogen: Etwa 90% (ca. 85 000 am 1.12.2013) der in der öffentlich zugänglichen Datenbank für Biomoleküle – der Protein Data Bank [1] (PDB) – enthaltenen Strukturen wurden durch Einkristall-Kristallografie gelöst, während über weitere 10 000 Strukturen NMR-spektroskopisch aufgeklärt wurden. 2006 erschien eine bemerkenswerte Veröffentlichung [2], die zeigte, dass einige der NMR-Strukturen signifikante Fehler enthalten. Dies nahmen Heiko Möller, damals Juniorprofessor für NMR-Spektroskopie an komplexen molekularen Systemen und heute Professor in Potsdam, und ich zum Anlass, um Methoden zur Evaluation dieser Strukturen zu entwickeln. Als ein naheliegendes Kriterium konnte der Vergleich von gemessenen und berechneten NMR-chemischen Verschiebungen identifiziert werden, da diese für die Zuordnung der auf dem Kern-Overhauser-Effekt basierenden (NOESY-Spektren) Abstandskriterien bestimmt werden müssen, aber dann für die eigentliche Strukturbestimmung nicht mehr herangezogen werden. Sie sind durch Datenbanken wie die Biological Magnetic Resonance Bank (BMRB) [3] auch leicht zugänglich. Weitere Vorteile bestehen darin, dass die chemische Verschiebung extrem von der Umgebung des betrachteten Kerns abhängt, also eine hochsensible Sonde für die lokale Atomanordnung ist, und es viele quantenchemische, aber auch empirische Methoden für ihre Berechnung gibt. Theorie hilft dem Experiment Empirische Methoden [4,5] haben den Vorteil, dass sie sehr schnell sind und auch für große Systeme wie Proteine und DNA/RNA angewendet werden können. Andererseits sind diese aber durch ihre Parametrisierung an bekannten Strukturen eher Mittelwerte über ähnliche Strukturen und können damit die Feinheiten einer gegebenen Struktur nicht exakt nachbilden. Quantenchemische Verfahren kommen ohne jegliche Parametrisierung aus und zeigen durch diese Ab-initio-Vorhersage wie erwartet eine deutlich stärkere Sensibilität gegenüber unterschiedlichen Bindungslängen, Bindungswinkeln und Abständen zu benachbarten Atomen. Standardverfahren können wegen der benötigten Rechenzeit nur für kleine Moleküle verwendet werden, sodass im Zusammenhang mit den für uns interessanten biochemischen Systemen z.B. auf Fragmentmethoden wie unseren Field-Adapted Adjustable Density Matrix Assembler (FA-ADMA) [6] zurückgegriffen werden muss. Dieser teilt das Gesamtsystem in Subsysteme, die dann einzeln berechnet und zum Schluss wieder zusammengesetzt werden. Dadurch wurde es möglich, z.B. alle chemischen Verschiebungen der leichten Kette von Dynein 2a zu berechnen [7], das als ein Beispiel in der oben erwähnten Publikation [2] verwendet wurde. Das erste Strukturmodell (PDB-Eintrag 1TGO) zeigte ein Monomer, das kurze Zeit später zu einem Dimer berichtigt wurde (PDB-Eintrag 1Z09 [8]). Die Evaluation der ersten Struktur mithilfe von chemischen Verschiebungen hätte diesen Irrtum direkt aufzeigen können.
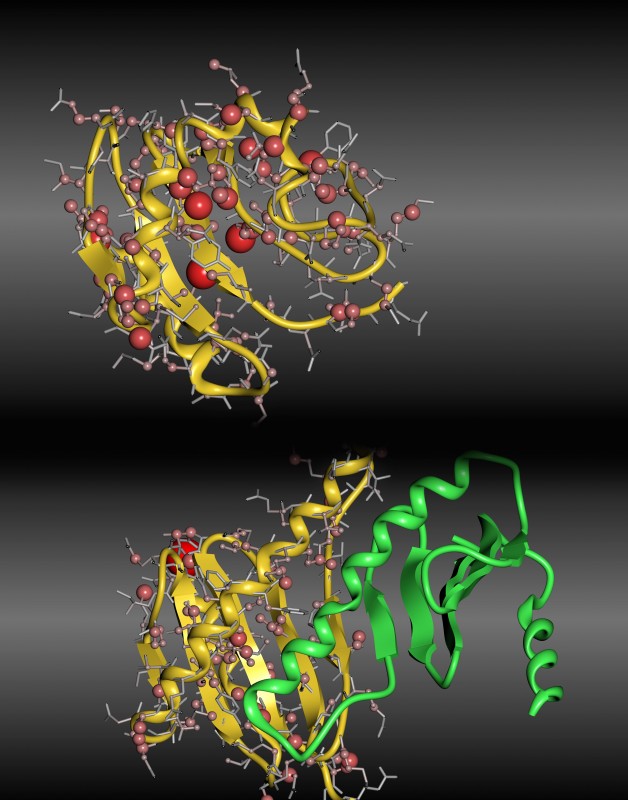
Abb.1 Unterschiede zwischen berechneten und gemessenen 13C-chemischen Verschiebungen für die leichte Kette von Dynein 2A, basierend auf der Monomer- (oben, 1TGO) bzw. Dimer-Struktur (unten, 1Z09). Die Abweichungen sind durch die Größe der Kugeln und die Farbintensität kodiert. Wie in Abbildung 1 zu sehen ist, zeigt die Monomerstruktur starke Abweichungen zwischen den berechneten und gemessenen 13C-chemischen Verschiebungen – und dies vor allem in der stark verformten α-Helix, die den zentralen Bereich des Monomers bildet. In der Dimer-Struktur bildet die α-Helix die Interaktionsfläche zwischen den beiden Monomeren und die chemischen Verschiebungen können fast optimal reproduziert werden. Nur ein Kern im hinteren Teil des Bildes zeigt eine größere Abweichung. Doch dieser liegt im N-Terminus der Struktur, dessen Flexibilität wie unten genauer gezeigt eine Vorhersage extrem erschwert. Eine Struktur ist nicht alles Für Protonen waren die mit diesem ersten Ansatz erhaltenen Ergebnisse deutlich schlechter und die Unterschiede konnten nicht mit Ungenauigkeiten in den Strukturen erklärt werden [7,9]. Betrachtet man die Art der schlecht vorhergesagten Kerne, deuten die polare Natur und die Nähe zur Oberfläche des Moleküls auf das Lösungsmittel als mögliche Erklärung hin. NMR-Messungen erfolgen in Lösung, im Fall von biochemischen Systemen im wässrigen Medium, wogegen die quantenchemischen Rechnungen im Vakuum bzw. mit einem impliziten Solvensmodell, in dem das Wasser durch ein Kontinuum mit einer Dielektrizitätskonstante von 78 beschrieben wird, durchgeführt werden. Beide Modelle sind nicht in der Lage, Wasserstoffbrücken zwischen dem betrachteten Molekül und dem Lösungsmittel zu beschreiben. Um diese Lösungsmitteleinflüsse in aller Tiefe zu untersuchen, entschieden wir uns für das kleinste Testsystem mit einer Peptidbindung und damit biologischer Relevanz [10]: das N-Methyl-Acetamid. Dieses zeigt im Experiment eine chemische Verschiebung des Amid-Protons HN von 7,74ppm und damit einen Unterschied zu den beiden Methylgruppen von 5,10 bzw. 5,84ppm (Um den Einfluss systematischer Fehler bei der Rechnung des verwendeten Standards Tetramethylsilan auszuschließen, werden hier im Weiteren immer die Unterschiede zu den beiden Methylgruppen verwendet.). Die Rechnung an einer minimierten Struktur im impliziten Lösungsmittel ergab aber nur Differenzen von 1,39 bzw. 2,61ppm und damit Fehler von über 3ppm. Dies entspricht genau dem Phänomen der zu niedrigen chemischen Verschiebungen polarer, solvensexponierter Protonen, die auch bei den Proteinen beobachtet wurde. Für eine bessere Beschreibung wurden Molekulardynamik-Simulationen durchgeführt, in denen das Molekül in expliziten Wassermolekülen gelöst vorlag. Aus dieser Simulation wurden dann einzelne Schnappschüsse entnommen und an diesen die NMR-Berechnungen durchgeführt. Neben der Verbesserung der Solvathüllenbeschreibung wird so auch die Flexibilität des Moleküls berücksichtigt. Für das HN wurden dabei chemische Verschiebungen in dem extrem großen Bereich zwischen 4 und 10ppm ermittelt. Den größten Einfluss hatte dabei der Abstand zum nächstgelegenen Wassermolekül, das eine Wasserstoffbrücke mit HN ausbildet (siehe Abb. 2).
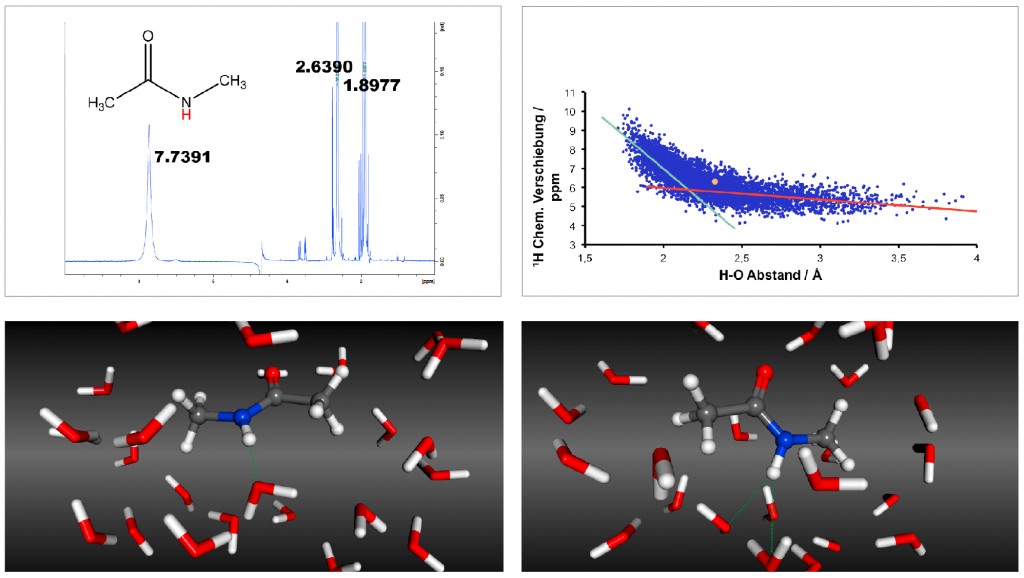
Abb.2 1H-chemische Verschiebungen von N-Methyl-Acetamid oben links: experimentelles Spektrum oben rechts: chemische Verschiebung des HN in Abhängigkeit vom Abstand zum nächsten Wassermolekül unten links: Schnappschuss mit sehr kleinem Abstand (1,79Å) und sehr hoher chemischer Verschiebung (10,12ppm) unten rechts: Schnappschuss mit sehr großem Abstand (2,49Å bzw. 3.00Å) und sehr niedriger chemischer Verschiebung (4.13ppm) Da die Zeitskala der NMR sehr langsam im Vergleich zur Simulation ist, sieht das Experiment den Mittelwert des thermodynamischen Ensembles, das hier durch die Schnappschüsse repräsentiert werden soll, und die Übereinstimmung verbessert sich deutlich mit Differenzen zu den Methylprotonen von 3,59 bzw. 4,47ppm bei Verwendung von klassischen MD-Simulationen [10] und sogar von 5,17 bzw. 5,87ppm, wenn Ab-initio-MD-Simulationen verwendet werden [11]. Damit ist der Fehler in den Rechnungen, basierend auf dem bestmöglichen Ensemble, auf unter 0,1ppm gefallen. Das Experiment hilft der Theorie Wie gerade gezeigt, können chemischen Verschiebungen hervorragend vorhergesagt werden, wenn Lösungsmitteleffekte und die Flexibilität des Moleküls exakt berücksichtigt werden. Leider sind die dafür im Moment nötigen Ab-initio-Simulationen nur für kleine Moleküle wie N-Methyl-Acetamid durchführbar. Deswegen wäre es wünschenswert, die in der klassischen MD verwendeten Kraftfelder weiter zu optimieren. Dabei kann man den Spieß jetzt auch umdrehen. Da wir gezeigt haben, dass das optimale Ensemble zur perfekten Übereinstimmung zwischen Experiment und Theorie führt, kann diese Übereinstimmung nun als Kriterium zur Evaluation von Kraftfeldparametern und zum Aufzeigen von Problemregionen herangezogen werden. Wir sind gerade dabei, dies für DNA-Kraftfelder, die im AMBER-Softwarepaket [12] enthalten sind, zu tun.
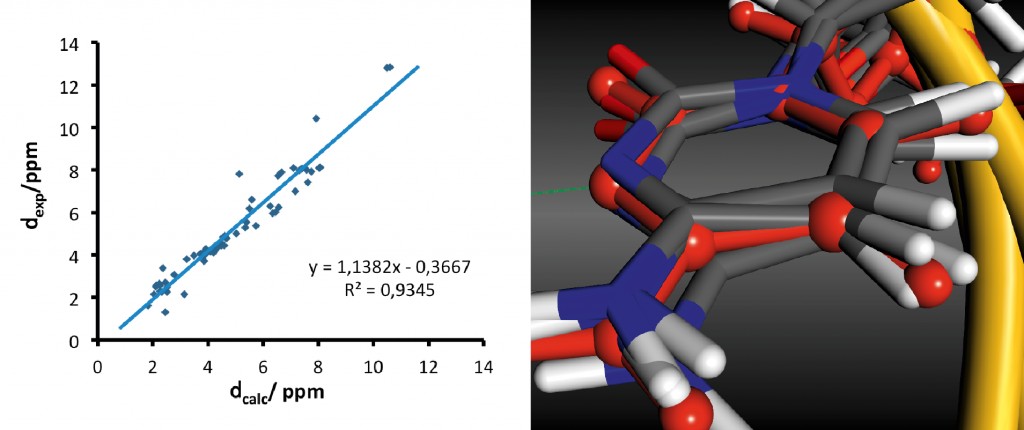
Abb.3 1H-chemische Verschiebungen der d(GCGAAGC)-Haarnadelstruktur (PDB-Eintrag 1KR8) [13] links: Korrelation zwischen berechneten und experimentellen Werten rechts: Überlagerung der experimentellen Struktur (rotes Kugel-und Stabmodell) und fünf Schnappschüssen aus der MD (atomfarbkodierte Stabmodelle) – deutlich ist der längere Abstand der Wasserstoffbrücke in den MD-Strukturen zu erkennen. Abbildung 3 zeigt die Korrelation für eine kleine Haarnadelstruktur [13], die bereits einen sehr guten Korrelationskoeffizienten aufweist und für die meisten Kerne nur geringe Fehler zeigt. Nur wenige deutliche Ausreißer sind sichtbar. Ein Beispiel dafür sind die H1-Protonen von 2 Guanin-Basen, die an Basenpaarungen beteiligt sind. Diese werden im Experiment mit ca. 13ppm bestimmt, aber nur mit knapp über 10ppm berechnet. Wie auch in der Abbildung zu sehen ist, ist dies auf die zu lange und zu schwache Wasserstoffbrücke zwischen den Basen zurückzuführen. Hier ergibt sich also Potenzial für ein verbessertes Kraftfeld, das auch nichtkanonische Strukturen beschreiben kann.
Literatur Foto: © panthermedia | Oleksii Lukin |
L&M 1 / 2014
Das komplette Heft zum kostenlosen Download finden Sie hier: zum Download Der Autor:Weitere Artikel online lesen


NewsSchnell und einfach die passende Trennsäule finden
© Text und Bild: Altmann Analytik ZEISS stellt neue Stereomikroskope vor
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen. © Text und Bild: Carl Zeiss Microscopy GmbH |





