|
Neueste Entwicklungen auf dem Gebiet der In-silico-Erforschung von Signalwegen
Neueste Entwicklungen auf dem Gebiet der In-silico-Erforschung von Signalwegen
Herkömmliche Computersimulationsmethoden können aufgrund ihres hohen Rechenaufwandes bisher nur zur Untersuchung des Frühstadiums von Signalwegen biologisch lebenswichtiger Signalproteine eingesetzt werden. Ihr Einsatzspektrum reicht daher typischerweise zur Erfassung von schnellen Prozessen im Bereich von Femtosekunden bis hin zu mehreren Mikrosekunden für Systeme in der Größenordnung von bis zu 100.000 Atomen. Die Entwicklung neuer ratenbasierter Kinetik-Monte-Carlo(KMC)-Methoden eröffnet neuerdings die Möglichkeit zur Studie der Mehrskalen-Relaxationsdynamik großskaliger enzymatisch-aktiver Signal-Protein-Systeme auf biologisch relevanten Zeitskalen im Bereich von Submillisekunden bis hin zu Sekunden mit relativ moderatem Rechenaufwand.
Signalproteine steuern zentrale biologische Prozesse in Zellen

Signalproteinkomplexe fungieren als Regulatoren der Signalwege von Zellen in lebenden Organismen, die auf eine Vielzahl von Umweltreizen wie z. B. Licht, Temperaturveränderung und/oder mechanische Belastung reagieren. Eine Klasse von Signalproteinen, die in Säugetieren von zentraler Bedeutung sind, sind die Rac-Proteine [1].
Es sind kleine GTPasen, die lebenswichtige Zellvorgänge wie z. B. Wachstum, Membran-Adhesion und das Überleben von Zellen steuern. Ihre Aktivierung wird durch Bindung eines spezifischen Effektorproteins an charakteristische Aktivierungsstellen der jeweiligen GTPase im Zellzyklus ausgelöst.
Zum Beispiel ist von der GTPase Rac1 bekannt, dass sie einen Komplex mit der Serin-Threonin-Protein-Kinase PAK1 an der sog. SwitchII-Aktivierungsstelle bildet. Außerdem, werden deren Mutation verdächtigt zur Deregulierung von Prozessen beizutragen, die bösartige Tumore hervorrufen. Signalproteine spielen auch eine wichtige Rolle in Pflanzen, in denen sie typischerweise auf eine Veränderung der Lichtintensität und/oder Temperatur reagieren. Ein Beispiel hierfür stellen die sog. Fototropine dar [2]. Es sind blaulichtempfindliche Proteinkomplexe, die eine große Vielfalt an biologischen Prozessen wie z. B. die Pflanzenbewegung zum oder aus dem Sonnenlicht (Fototropismus) in höheren Pflanzen oder Mikroalgen regulieren. Sie bestehen aus 2 Light-Oxygen-Voltage-(LOV)-sensitiven Protein-Domänen, welche jeweils ein nichtkovalent gebundenes Flavin-Mono-Nukleotid-(FMN)-Chromophor beinhalten, und einer C-terminalen Serin-Threonin-Kinase (siehe Abb. 2). Durch Absorption von
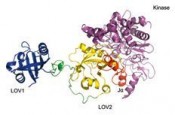 blauem Licht wird eine kovalente Bindung zwischen dem FMN-Chromophor und einem angrenzenden reaktiven Cystein des Apo-Proteins gebildet. Dieser Vorgang führt anschließend die Aktivierung der Kinase herbei, die ein Signal an den Organismus über die Auto-Phosporylierung des Fototropins aussendet. Während die In-vivo-Funktionalität der LOV1-Domäne innerhalb des Proteinkomplexes immer noch unklar ist, wurde bereits herausgefunden, dass die fotochemische Reaktivität der LOV2-Domäne von zentraler Bedeutung für die Aktivierung der Kinase ist. Wie mittels einer Serie von experimentellen Untersuchungen gezeigt wurde, verliert die LOV2-Domäne unter Lichteinwirkung ihren inhibitorischen Charakter auf die Kinase durch Abspaltung einer peripheren a-Helix, die sog. Ja-Helix, vom Kern der LOV-Domäne. Das Frühstadium des Aktivierungsmechanismus wurde kürzlich mit atomarer Auflösung mithilfe von Computersimulationen in unserer Arbeitsgruppe aufgeklärt [3]. Durch Verknüpfung des AsLOV2-Ja-Fotoschalters und der vorhin erwähnten Rac1-GTPase konnten Wu et al. [4] vor Kurzem ein genetisch codiertes Fusionsprotein, die sog. fotoaktivierbare Rac1-GTPase (PA-Rac1), erzeugen, welche sich denselben Fotoschaltungsmechanismus zur Modulation des Signalverhaltens von Rac1 zu Nutze macht. Insbesondere konnten sie nachweisen, dass im Dunkelzustand die AsLOV2-Domäne des PARac1-Konstrukts die Rac1-GTPase durch Blockieren seiner Wechselwirkung zum
blauem Licht wird eine kovalente Bindung zwischen dem FMN-Chromophor und einem angrenzenden reaktiven Cystein des Apo-Proteins gebildet. Dieser Vorgang führt anschließend die Aktivierung der Kinase herbei, die ein Signal an den Organismus über die Auto-Phosporylierung des Fototropins aussendet. Während die In-vivo-Funktionalität der LOV1-Domäne innerhalb des Proteinkomplexes immer noch unklar ist, wurde bereits herausgefunden, dass die fotochemische Reaktivität der LOV2-Domäne von zentraler Bedeutung für die Aktivierung der Kinase ist. Wie mittels einer Serie von experimentellen Untersuchungen gezeigt wurde, verliert die LOV2-Domäne unter Lichteinwirkung ihren inhibitorischen Charakter auf die Kinase durch Abspaltung einer peripheren a-Helix, die sog. Ja-Helix, vom Kern der LOV-Domäne. Das Frühstadium des Aktivierungsmechanismus wurde kürzlich mit atomarer Auflösung mithilfe von Computersimulationen in unserer Arbeitsgruppe aufgeklärt [3]. Durch Verknüpfung des AsLOV2-Ja-Fotoschalters und der vorhin erwähnten Rac1-GTPase konnten Wu et al. [4] vor Kurzem ein genetisch codiertes Fusionsprotein, die sog. fotoaktivierbare Rac1-GTPase (PA-Rac1), erzeugen, welche sich denselben Fotoschaltungsmechanismus zur Modulation des Signalverhaltens von Rac1 zu Nutze macht. Insbesondere konnten sie nachweisen, dass im Dunkelzustand die AsLOV2-Domäne des PARac1-Konstrukts die Rac1-GTPase durch Blockieren seiner Wechselwirkung zum
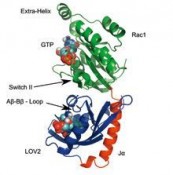 Effektorprotein PAK1 inhibiert. Dagegen wird im Lichtzustand die sterische Inhibition durch Abspaltung der Ja-Helix vom LOV-Proteinkern beseitigt, was die Aktivierung der Rac1-GTPase ermöglicht (siehe Abb. 3). Durch Expression des PA-Rac1's in Krebszellen des HeLa-Typs konnten sie außerdem demonstrieren, dass dieses Proteinkonstrukt zur Kontrolle der Zellbeweglichkeit und Zellfunktionalität über Lichtpulse eingesetzt werden kann.
Effektorprotein PAK1 inhibiert. Dagegen wird im Lichtzustand die sterische Inhibition durch Abspaltung der Ja-Helix vom LOV-Proteinkern beseitigt, was die Aktivierung der Rac1-GTPase ermöglicht (siehe Abb. 3). Durch Expression des PA-Rac1's in Krebszellen des HeLa-Typs konnten sie außerdem demonstrieren, dass dieses Proteinkonstrukt zur Kontrolle der Zellbeweglichkeit und Zellfunktionalität über Lichtpulse eingesetzt werden kann.
Untersuchung des Frühstadiums der Signalwege von Signalproteinen mit herkömmlichen Computersimulationsmethoden
Um die Strukturdynamik von solchen Proteinkomplexen auf atomarer Längenskala zu beschreiben, wurden mehrere Rechenmethoden, beginnend in den späten 70er-Jahren, entwickelt. Ein prominentes Beispiel unter ihnen ist die MD-Technik, die die Zeitentwicklung von Vielteilchensystemen durch den Phasenraum über die numerische Integration der Newton'schen Bewegungsgleichungen beschreibt. Da sich jedoch ihr Anwendungsspektrum für kleine Proteinsysteme typischerweise nur von Nanosekunden bis hin zu Submikrosekunden erstreckt, ist ihr Nutzen zur Studie von Signalprozessen auf den üblichen experimentellen Zeitskalen eher begrenzt. Um längere Zeitskalen mit der MD-Technik zu erreichen, wurden in den letzten Jahrzehnten mehrere Methoden vorgeschlagen. Eine von ihnen ist die Coarse-Graining(CG)-Methode, in der die Anzahl von Wechselwirkungen durch Reduktion der Systemfreiheitsgrade verringert wird. Dies ermöglicht es wiederum, den Rechenaufwand durch Einsatz größerer Zeitschritte zu erniedrigen. Eine erfolgreiche CG-Methode für Proteinsysteme ist die United-Atom-Methode, die z. B. im GROMOS96-Kraftfeld [5] implementiert ist.
Ihr Konzept besteht darin, dass alle Wasserstoff-Atome mit ihren dazugehörigen aliphatischen Kohlenstoffatomen jeweils als eine einzige effektive atomare Einheit dargestellt werden. Eine solche Methode wurde erst kürzlich von uns zur Aufklärung des Frühstadiums der Aktivierung des PA-Rac1-Photoenzyms mit atomarer Auflösung eingesetzt [6]. Methoden, die ein noch stärkeres Coarse-Graining durchführen wie z. B. die MARTINI-Methode [7], reproduzieren zuverlässig die Proteinstruktur. Sie versagen aber in der Wiedergabe der korrekten Dynamik des komplexen Proteinsystems aufgrund der stärkeren Heterogenität der Proteinstruktur auf atomarer Beschreibungsebene.
Darüber hinaus ist ein weiteres Problem ihre schlechte Übertragbarkeit, da sie spezifisch an die Natur des betrachteten physikalischen Problems angepasst sind. Daher sind sie meist nicht geeignet, die Mehrskalen-Relaxationsdynamik von komplexen Proteinsystemen weit entfernt vom Gleichgewicht zu reproduzieren.
Um die Reichweite von Computersimulationstechniken auf ausgeprägte Nichtgleichgewichtssituationen zu erweitern, wurden Multiple-Timestepping-Methoden wie z. B. die Reversible-Reference-System-Propagator-Algorithmen (RESPA’s) [8] entwickelt, die die Simulation von Proteinsystemen durch eine Trennung von Zeitskalen und/oder Potenzialbereichen erheblich beschleunigen. Sie beruhen im Allgemeinen auf einer Zerlegung des dynamischen Spektrums des Makromoleküls in langsame und schnelle Moden und erlauben eine effizientere Propagation der langsamen dynamischen Komponenten durch Verwendung größerer Zeitschritte im numerischen Integrationsverfahren. Mit diesen Methoden konnte eine Beschleunigung von 10- bis zu 100-mal im Vergleich zu konventionellen MD-Techniken erzielt werden, was zu einer Ausdehnung der Anwendbarkeit von MD-Techniken auf den Mikrosekunden- oder gar Submillisekundenbereich für Peptide und kleine Proteine führte. Auch wenn in diesen Fällen eine substanzielle Erweiterung des Spektrums der rechnerisch erfassbaren Zeitskalen erreicht werden konnte, erfordern sie zum Studium des Langzeitverhaltens von komplexen Signalproteinen dennoch einen erheblichen Rechenaufwand. Dies bedeutet in der Regel, dass die Simulationsläufe mit solchen Methoden mehrere Monate auf parallelen Computerclustern oder Supercomputern benötigen, was ihre Brauchbarkeit für solche Anwendungen bis in die absehbare Zukunft erheblich einschränkt. Andere Techniken wie z. B. die Meta-Dynamik-Methode [9] ermöglichen ein schnelleres Sampling der freien Energiehyperfläche von Peptiden und mittelgroßen Proteinen unter Verwendung von modifizierten Potenzialen bzw. effektiven Hamilton- Funktionen. Sie erlauben eine signifikante Ersparnis in Bezug auf den Rechenaufwand im Vergleich zu MD-basierten Methoden. Jedoch sind sie aufgrund ihrer Natur und Eigenschaft grundsätzlich nicht geeignet, die reale Dynamik von komplexen Proteinsystemen zu beschreiben.
Erfassung der Mehrskalen-Signal-Transduktionsdynamik von Proteinen auf biologisch relevanten Zeitskalen durch neue, gekoppelte KMC-MD-Methoden
Während herkömmliche, MD-basierte Simulationstechniken ohne Weiteres zur Untersuchung des Frühstadiums der Signalwege komplexer Proteinsysteme bei atomarer Auflösung eingesetzt werden können, ist ihr Nutzen zur Aufklärung der Mehrskalen-Transduktionsdynamik der meisten Signalprozesse auf biologisch relevanten Zeitskalen aufgrund ihres hohen Rechenaufwands, eher begrenzt. Dies liegt darin begründet, dass sich ihre Signalwege von der Relaxation der Aminosäuren im Bereich von Femtosekunden- bzw. Pikosekunden bis hin zur Relaxation der Sekundär- bzw. Tertiärstrukturelemente im Sekundenbereich erstrecken. Zur Erfassung solcher Prozesse haben wir kürzlich eine neue
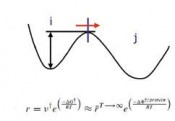 Mehrskalen-Modellierungsmethode entwickelt, bei der die KMC-Methode mit einem MD-Algorithmus verknüpft wurde. Die grundlegende Idee dieser sog. KMC-MD-Methode beruht darauf, dass der Signalweg großer Proteinkomplexe durch seltene Ereignisse dominiert wird, d.h., die Relaxationsdynamik wird durch gelegentliche Übergänge von einem Zustand in einen anderen Zustand mit langen Phasen relativer Inaktivität zwischen den Übergängen bestimmt. Die letzteren Phasen entstehen deswegen, weil die Simulationstrajektorie die Energiebarrieren überwinden muss, um von einem Zustand in den anderen zu gelangen (siehe Abb. 4). Sie gewährleisten, dass die Überschussenergie des Systems an die Umgebung abgegeben werden kann, was die Relaxation des Systems ermöglicht, und dass die Konfigurationen statistisch unabhängig voneinander sind. Diese Phasen können mithilfe eines MD-Algorithmus simuliert werden. Die Raten aller möglichen Übergänge aus jedem Zustand werden mithilfe eines biomolekularen Kraftfeldes am Ende jeder MD-Relaxationsphase berechnet und die wahrscheinlichsten Übergänge mithilfe eines KMC-Algorithmus ausgewählt. Hierbei wird die Annahme getroffen, dass der Signalweg durch charakteristische geschwindigkeitsbestimmende Prozesse wie z.B. Bruch und Bildung von H-Brücken bestimmt wird. Hieraus ergeben sich verschiedene Simulationsphasen, in denen die Dynamiken verschiedener Relaxationseinheiten wie z. B. der Residuen oder Sekundärstrukturelemente zur Mehrskalen-Transduktionsdynamik beitragen. Da diese Übergänge nur selten stattfinden, kann mithilfe des KMC-Algorithmus eine sehr große Beschleunigung der Simulation und Verringerung des Rechenaufwandes im Vergleich zur konventionellen MD-Methode erzielt werden. Um ihre Zuverlässigkeit und Effizienz zu testen, haben wir die KMC-MD-Methode zur Untersuchung der Signalwege komplexer LOV-Protein-Systeme des Wildtyps und deren Mutanten wie z.B. den AsLOV2-J?-Fotoschalter, PA-Rac1-GTPase und Fototropin eingesetzt. Dabei konnten wir sowohl experimentelle Beobachtungen bestätigen als auch zentrale neue Erkenntnisse über diese Proteinsysteme gewinnen. Wir haben herausgefunden, dass nach Fotoanregung mit blauem Licht diese Systeme typischerweise einen komplexen Signalweg unter Ausführung einer Spannungsrelaxationsdynamik über mehrere Längen- und Zeitskalen durchlaufen. Dies impliziert zunächst lokale strukturelle Veränderungen auf der Ebene der Residuen in der Nähe des lichtempfindlichen Reaktionszentrums im Zeitbereich von Nanosekunden, die wiederum extensive Umlagerungen der peripheren Proteinstrukturelemente auf Zeitskalen von mehreren Mikrosekunden bis hin zu Sekunden hervorrufen. Insbesondere haben wir mit unserer neuen Methode beobachtet, dass das Frühstadium des Signalweges durch Kopplung der Ib- und Hb-Stränge über H-Brückenbildung zwischen einem Glutamin und Asparagin in der Nähe des FMN's (Gln513 und Asn492) charakterisiert wird.
Mehrskalen-Modellierungsmethode entwickelt, bei der die KMC-Methode mit einem MD-Algorithmus verknüpft wurde. Die grundlegende Idee dieser sog. KMC-MD-Methode beruht darauf, dass der Signalweg großer Proteinkomplexe durch seltene Ereignisse dominiert wird, d.h., die Relaxationsdynamik wird durch gelegentliche Übergänge von einem Zustand in einen anderen Zustand mit langen Phasen relativer Inaktivität zwischen den Übergängen bestimmt. Die letzteren Phasen entstehen deswegen, weil die Simulationstrajektorie die Energiebarrieren überwinden muss, um von einem Zustand in den anderen zu gelangen (siehe Abb. 4). Sie gewährleisten, dass die Überschussenergie des Systems an die Umgebung abgegeben werden kann, was die Relaxation des Systems ermöglicht, und dass die Konfigurationen statistisch unabhängig voneinander sind. Diese Phasen können mithilfe eines MD-Algorithmus simuliert werden. Die Raten aller möglichen Übergänge aus jedem Zustand werden mithilfe eines biomolekularen Kraftfeldes am Ende jeder MD-Relaxationsphase berechnet und die wahrscheinlichsten Übergänge mithilfe eines KMC-Algorithmus ausgewählt. Hierbei wird die Annahme getroffen, dass der Signalweg durch charakteristische geschwindigkeitsbestimmende Prozesse wie z.B. Bruch und Bildung von H-Brücken bestimmt wird. Hieraus ergeben sich verschiedene Simulationsphasen, in denen die Dynamiken verschiedener Relaxationseinheiten wie z. B. der Residuen oder Sekundärstrukturelemente zur Mehrskalen-Transduktionsdynamik beitragen. Da diese Übergänge nur selten stattfinden, kann mithilfe des KMC-Algorithmus eine sehr große Beschleunigung der Simulation und Verringerung des Rechenaufwandes im Vergleich zur konventionellen MD-Methode erzielt werden. Um ihre Zuverlässigkeit und Effizienz zu testen, haben wir die KMC-MD-Methode zur Untersuchung der Signalwege komplexer LOV-Protein-Systeme des Wildtyps und deren Mutanten wie z.B. den AsLOV2-J?-Fotoschalter, PA-Rac1-GTPase und Fototropin eingesetzt. Dabei konnten wir sowohl experimentelle Beobachtungen bestätigen als auch zentrale neue Erkenntnisse über diese Proteinsysteme gewinnen. Wir haben herausgefunden, dass nach Fotoanregung mit blauem Licht diese Systeme typischerweise einen komplexen Signalweg unter Ausführung einer Spannungsrelaxationsdynamik über mehrere Längen- und Zeitskalen durchlaufen. Dies impliziert zunächst lokale strukturelle Veränderungen auf der Ebene der Residuen in der Nähe des lichtempfindlichen Reaktionszentrums im Zeitbereich von Nanosekunden, die wiederum extensive Umlagerungen der peripheren Proteinstrukturelemente auf Zeitskalen von mehreren Mikrosekunden bis hin zu Sekunden hervorrufen. Insbesondere haben wir mit unserer neuen Methode beobachtet, dass das Frühstadium des Signalweges durch Kopplung der Ib- und Hb-Stränge über H-Brückenbildung zwischen einem Glutamin und Asparagin in der Nähe des FMN's (Gln513 und Asn492) charakterisiert wird.
Dieses Ereignis führt zu einer N-terminalen Entfaltung der Ja-Helix und ihrer nachfolgenden Abspaltung vom LOV-Proteinkern. Im Falle des PA-Rac1-Systems konnten wir zeigen, dass der letztere Abspaltungsprozess zur Freisetzung einer funktionellen a-Helix an der SwitchII-Aktivierungsstelle der Rac1-GTPase führt. Diese ist als primäre Bindungsstelle von Rac1 mit seinen Effektor- Protein-Domänen bekannt, wie hier am Beispiel von PAK1 gezeigt (siehe Abb. 6). Dagegen konnten im Falle des Dunkelzustandes sowohl des AsLOV2-Ja-Fotoschalters als auch der PA-Rac1-GTPase keine Aktivierung festgestellt werden. Durch Gegenüberstellung dieser Ergebnisse mit den derzeit verfügbaren experimentellen Ergebnissen des LOV-Wildtyps und verschiedener Mutanten konnten wir schlussfolgern, dass unsere Methode eine neue zuverlässige
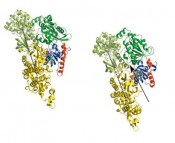 Modellierungstechnik zur Simulation des Mehrskalen-Signalrelaxationsverhaltens großer Proteinkomplexe darstellt. Darüber hinaus konnten wir die Gesamtdauer der Simulationsläufe herkömmlicher kraftfeldbasierter Computersimulationsmethoden auf typische Zeitskalen, die in der Natur oder in biologischen Experimenten im Zeitraum von Mikrosekunden bis hin zu Sekunden ablaufen, ausdehnen. Die Essenz der KMC-MD-Methode wurde kürzlich in der Fachzeitschrift Journal of Chemical Physics [10] veröffentlicht.
Modellierungstechnik zur Simulation des Mehrskalen-Signalrelaxationsverhaltens großer Proteinkomplexe darstellt. Darüber hinaus konnten wir die Gesamtdauer der Simulationsläufe herkömmlicher kraftfeldbasierter Computersimulationsmethoden auf typische Zeitskalen, die in der Natur oder in biologischen Experimenten im Zeitraum von Mikrosekunden bis hin zu Sekunden ablaufen, ausdehnen. Die Essenz der KMC-MD-Methode wurde kürzlich in der Fachzeitschrift Journal of Chemical Physics [10] veröffentlicht.
Rechenaufwand der gekoppelten KMC-MD-Methode im Vergleich zur herkömmlichen MD-Methode
Abschließend geben wir noch eine Abschätzung des Rechenaufwandes, der mithilfe der KMC-MD-Methode im Vergleich zur herkömmlichen MD-Methode erforderlich ist, um ähnliche Zeitskalen zu erreichen. Hierzu betrachten wir den Lichtzustand des PA-Rac1-Systems, für welchen eine Simulationsgesamtdauer von 300 ?s unter Verwendung der KMC-MD-Methode erzielt wurde. In dieser Berechnung wurde eine Gesamtzahl von 40 KMC-Schritten durchgeführt, die eine totale Anzahl an 200000 MD-Relaxationsschritten benötigten.
Wenn wir nun die konventionelle MD-Simulationstechnik mit einem Zeitschritt von 1 fs benutzen würden, würden wir eine Gesamtzahl von 3 * 1000000000000 MD-Schritten benötigen, um die gleiche Zeit von 300 ?s wie mit der KMC-MD-Methode zu erreichen. Unter Berücksichtigung, dass die Kosten für die Ausführung der KMCEreignisse in der Größenordnung einer MD-Relaxationsphase liegen, schlussfolgern wir, dass die KMC-MD-Simulationsmethode den Rechenaufwand um den Faktor 7.5 * 1000000 für den Fall des PA-Rac1-Systems im Lichtzustand verringert. Weitere Gewinne in der Recheneffizienz können mithilfe von Multiple-Timestepping-Methoden wie z. B. RESPA-Algorithmen [8] zur Beschleunigung der Berechnung der MD-Relaxationsphasen erzielt werden. Schließlich ist noch wichtig zu betonen, dass der zu Grunde liegende Algorithmus auf eine große Anzahl an alternativen Protein-Solvenz-Systemen übertragen werden kann und neue Wege zur Untersuchung der lichtinduzierten Regulation enzymatischer Reaktivität sowie von Genfunktionen eröffnen kann.
Für eine Ribbon Darstellung des Proteinkomplexes folgen Sie einfach diesem Link!
Literatur
[1] A. Y. Chan, S. J. Coniglio, Y.-Y. Chuang, D. Michaelson, U. G. Knaus, M. R. Philips und M. Symons (2005) Oncogene 24, 7821 –7829.
[2] P. Hegemann (2008) Annu. Rev. Plant. Biol. 59, 167 –189.
[3] E. Peter, B. Dick und S.A. Baeurle (2010)Nat. Commun. 1, 122.
[4] Y. I. Wu, D. Frey, O. I. Lungu, A. Jaehrig, I. Schlichting,B. Kuhlman und K. M. Hahn (2009) Nature 461, 104 –108.
[5] E. Lindahl, B. Hess und D. van der Spoel (2001) J. Mol. Model. 7, 306 –317.
[6] E. Peter, B. Dick und S. A. Baeurle (2012) Prot. Struct. Funct. Bioinf. 80, 1350 –1362
[7] L. Monticelli, S. K. Kandasamy, X. Periole, R. G. Larson, D. P. Tieleman und S. J. Marrink (2008) J. Chem. Theory Comput. 4, 819 –834.
[8] M. Tuckerman, B. J. Berne und G. J. Martyna (1992) J. Chem. Phys. 97, 1990 –2001
[9] A. Barducci, R. Chelli, P. Procacci, V. Schettino, F. L. Gervasio und M. Parrinello (2006) J. Am. Chem. Soc. 128, 2705 –2710.
[10] E. Peter, B. Dick und S. A. Baeurle (2012) J. Chem. Phys. 136, 124112.
Abb1: © Jens Gimmler, MOLCAD GmbH
Abb2-5: © PD Dr. Stephan A. Baeurle
|
L&M 5 / 2012
Diese Artikel wurden veröffentlicht in Ausgabe L&M 5 / 2012.
Das komplette Heft zum kostenlosen Download finden Sie hier:
zum Download
Der Autor:
Weitere Artikel online lesen
News
Mit dem HPLC-Säulenkonfigurator unter www.analytics-shop.com können Sie stets die passende Säule für jedes Trennproblem finden. Dank innovativer Filtermöglichkeiten können Sie in Sekundenschnelle nach gewünschtem Durchmesser, Länge, Porengröße, Säulenbezeichnung u.v.m. selektieren. So erhalten Sie aus über 70.000 verschiedenen HPLC-Säulen das passende Ergebnis für Ihre Anwendung und können zwischen allen gängigen Herstellern wie Agilent, Waters, ThermoScientific, Merck, Sigma-Aldrich, Chiral, Macherey-Nagel u.v.a. wählen. Ergänzend stehen Ihnen die HPLC-Experten von Altmann Analytik beratend zur Seite – testen Sie jetzt den kostenlosen HPLC-Säulenkonfigurator!
© Text und Bild: Altmann Analytik
Aufnahme, Dokumentation und Teilen von Ergebnissen mit ZEISS Stemi 305 und ZEISS Stemi 508
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen.
© Text und Bild: Carl Zeiss Microscopy GmbH
|










