Calcium Imaging
Dynamisches Imaging von Calcium und STIM1 in ein und derselben Zelle mit Weitfeld- und TIRF-Mikroskopie
Simon Walker1, Nicholas Cunniffe1, Martin Bootman1, H. Llewelyn Roderick1,2
1 The Babraham Institute, Laboratory of Molecular Signalling, Cambridge, Großbritannien
2 Department of Pharmacology, University of Cambridge, Cambridge, Großbritannien
Ob im Leben oder Sterben – ohne Calcium geht in Zellen und im Gewebe fast gar nichts. Zellen benötigen Calcium, zum Beispiel um sich zu teilen und fortzubewegen. Nervenzellen brauchen es für die Signalübertragung und damit für den Informationsfluss zum und vom Gehirn. Auch für die Kontraktion von Herzzellen ist Calcium erforderlich. Für so vielfältige Einsätze in den unterschiedlichsten Zellen sind im Laufe der Zeit zahlreiche Proteine entstanden, mit deren Hilfe die Zellen die Calcium-Signale entschlüsseln und entsprechend darauf reagieren können.

Wie das im Einzelnen funktioniert, liegt auch heute noch weitestgehend im Dunkeln. Ein genaues Verständnis für diese vielfältigen biochemischen Prozesse innerhalb lebender Zellen ist aber nicht nur wichtig, um den lebenden Organismus als Ganzes genauer kennenzulernen. Es ist auch von entscheidender Bedeutung, wenn wir wissen wollen, wann und warum Zellprozesse aufhören zu funktionieren und dadurch Krankheiten verursachen. Innovative Technologien haben zu zahlreichen neuen Erkenntnissen in der Molekularbiologie beigetragen. Viele Fragen hinsichtlich der Struktur und Funktion von Zellen lassen sich aber nur durch das Beobachten lebender Zellen beantworten. STIMulierende Moleküle Der Eintritt von Calcium-Ionen (Ca2+) durch die Plasmamembran ist einer der Schlüsselprozesse für die Regulierung der intrazellulären Ca2+-Konzentration. Er wird durch die rezeptorvermittelte Freigabe von Calcium aus intrazellulären Speichern im endoplasmatischen Retikulum (ER) aktiviert. Dieser speicherabhängige Calcium-Einstrom (store-operated calcium entry/SOCE) ist ein gängiger Mechanismus für die Auffüllung interner Calcium- Speicher in vielen Zelltypen [1].
Das Calcium, das auf diesem Wege transportiert wird, wird häufig als CRAC (Calcium- Release Activated Current) bezeichnet und strömt über CRAC- oder SOCE-Kanäle in der Plasmamembran ein [2]. Der CRAC-Kanal ist der bestbeschriebene speicherabhängige (store-operated calcium/SOC) Einstromkanal und von entscheidender Bedeutung für die Immunreaktion. Die ununterbrochene Aktivität der CRAC-Kanäle ist für die Genexpression und Proliferation erforderlich. Sowohl die porenbildende Untereinheit des CRAC-Kanals als auch das Protein, das den Füllstand des ER misst, um den Calcium-Einstrom zu aktivieren, wurden kürzlich beschrieben [2]. Über die Kopplungsanalyse mit Proben von Patienten mit schwerer Immuninsuffizienz und RNAi- Screens konnte ORAI1 als porenbildende Untereinheit von CRAC identifiziert werden [3]. Das Protein, das eine Kopplung zwischen Füllstand des ER-Speichers und Calcium- Einstrom via ORAI1-Aktivierung herstellt, wurde als STIM1 (Stromal Interaction Molecule 1) identifiziert. STIM1 ist ein Membranphosphoprotein mit einer Transmembranhelix, das über eine Calcium-bindende EF-Hand in seiner dem ER-Lumen zugewandten Domäne verfügt. Es fungiert damit als Ca2+-Sensor. Nach der Freisetzung gespeicherten Calciums registrieren die vorher diffus in der ER-Membran verteilten STIM1-Proteine die interne Speicherentleerung auf und sammeln sich an der Plasmamembran. Hier beeinflussen sie die Aktivität verschiedener Calcium-Kanäle – darunter SOC und CRAC – und stellen eine entscheidende Verbindung mit den internen Calcium-Speichern her [4]. Eine STIM Isoform, STIM2, wurde ebenfalls identifiziert. Mittels einer EF- Hand mit geringerer Calcium-Sensitivität reguliert es den basalen Einstrom von Calcium [5].
Dynamisches Imaging von Calcium und STIM1
Die Autoren (Kontaktangaben siehe oben) konnten mithilfe von Lebendzell-Imaging intrazelluläres Calcium zusammen mit zwei verschiedenen Fluoreszenzproteinen in ein und derselben Zelle nahezu gleichzeitig beobachten. Der vorliegende Artikel beschreibt, wie sie hochentwickeltes Weitfeld-Fluoreszenz-Imaging mit Total Internal Reflection Fluorescence Microscopy (TIRFM) für Lebendzell-Mikroskopie kombinierten. Dieser Fluoreszenz-Imaging- Ansatz erlaubt die schnelle multidimensionale Analyse fluoreszenzmarkierter Zellen und damit das dynamische Imaging von Calcium und STIM1 innerhalb derselben Zelle.
Lebendzell-Imaging
Lebendzell-Imaging ermöglicht es, dynamische biologische Prozesse in lebenden Zellen in Echtzeit festzuhalten. Nie zuvor war innerhalb der Biochemie etwas Vergleichbares möglich. Die Entwicklung einer großen Bandbreite an spezifischen Fluoreszenzsonden für die nicht-invasive Untersuchung lebender Zellen, wie zum Beispiel Färbemittel und Fluoreszenz-Proteine, mit denen bestimmte Proteine markiert werden können, hat die Erforschung komplexer zellulärer Prozesse erleichtert. Durch die Entwicklung neuer Mikroskopie-Technologien sowie leistungsfähiger Soft- und Hardware für die digitale Bildverarbeitung und -analyse stellt das Lebendzell-Imaging heute eine Labormethode dar, die auch in Routineanwendungen in zahlreichen biomedizinischen Forschungsdisziplinen angewandt wird. Das Resultat sind immer mehr Studien, die auf zellulärer und subzellulärer Ebene Lebendzell-Mikroskopie und Imaging- Techniken einsetzen. Die Mikroskopie hat sich weiterentwickelt: weg von der rein strukturellen Charakterisierung fixierter Zellen, hin zur Erforschung von Prozessen in lebenden Zellen. Die statische morphologische Beobachtung kann jetzt durch die Charakterisierung dreidimensionaler (3-D) Architekturen zellulärer Strukturen ergänzt werden. Diese Innovationen erlauben es, dynamische zelluläre Aktivitäten im lebenden Gewebe mit Sub-Mikrometer-Auflösung und in Echtzeit zu beobachten. Darüber hinaus ermöglichen sie entscheidende Einblicke in die fundamentale Natur von Zell- und Gewebefunktionen, die mit fi xierten Zell-Techniken nicht möglich sind. Für solche Experimente mit lebenden Zellen müssen physiologische Bedingungen erfüllt sein – was durch die Fortschritte innerhalb der Mikroskopie-Technik in den letzten 20 Jahren enorm vereinfacht wurde. Mit der Erfindung der konfokalen Laser-Scanning-Mikroskopie sowie neuerer Entwicklungen wie die konfokale Spinning-Disk-Mikroskopie und TIRFM haben Lebendzell-Imaging-Systeme die Möglichkeit, biologische Prozesse zu erforschen, entscheidend verbessert. Darüber hinaus bieten sie die für das Imaging lebender Zellen erforderliche ho he räumliche Auflösung, ohne die Zellen dabei zu zerstören. Die hochentwickelten, modularen Fluoreszenz-Imaging- Stationen bieten spezielle „All-in-one“-Beleuchtungssysteme mit hochsensitiven Digitalkameras und präzisen Hardware-Kontrollboards und können vollständig in inverse wie aufrechte Mikroskope integriert werden. Diese Systeme wurden speziell für die Anforderungen experimenteller Anwendungen entwickelt und sind so optimiert, dass sie auch sehr schnelle Prozesse in lebenden Zellen festhalten können. Wissenschaftler sind dadurch in der Lage, komplexe Multiparameter-Protokolle für die simultane Erfassung und Interpretation zeitabhängiger Imaging-Daten zu entwickeln. Weitere Vorteile gegenüber früheren konfokalen und multiphotonen Mikroskopie-Instrumenten sind beispielsweise ultraschnelles Umschalten der Wellenlänge und Intensität. Spezielle Echtzeit-Controller koordinieren alle Hardware- und Peripherie-Komponenten und arbeiten unabhängig vom Imaging-Computer. Dies ermöglicht die präzise Synchronisation von Probenbeleuchtung und Echtzeit-Bildakquisition, wodurch Phototoxizität und Photobleaching minimiert werden.
In neuem Licht betrachtet
Neben den vielen optischen Hilfsmitteln zum Erfassen dieser zellulären und molekularen Vorgänge, eröffnet die TIRFM weitere bedeutende Optionen. So ist mithilfe der TIRFM eine totale Reflexion der Laserbeleuchtung im Glas des Objektträgers oder Gefäßes unterhalb der Probe möglich. Dadurch wird eine evaneszente (abklingende) Welle erzeugt, die mit einer geringen Tiefe in die Probenoberfläche eindringt und somit das Imaging von Fluoreszenz- markierten Molekülen in dieser Schicht bei hohem Signal-Rausch-Verhältnis (SRV) zulässt. Im Grunde genommen handelt es sich bei der TIRFM um Kontaktmikroskopie, die es Wissenschaftlern ermöglicht, speziell die Regionen, in denen Zellen an Nachbarzellen oder Substraten haften, deutlich zu visualisieren. TIRFM lässt einzelne Zellstrukturen und -prozesse in einem völlig neuen Licht erscheinen, wobei sich bereits etablierte Zellpräparations- und Verarbeitungsmethoden weiterhin ganz normal einsetzen lassen. Darüber hinaus ist die TIRFM einfach anzuwenden und überaus flexibel. Außerdem lassen sich die mit ihr generierten Ergebnisse schnell auswerten. Für eine leichtere Interpretation des sehr charakteristischen – und zunächst ungewohnten – Bildes einer Zelle in der TIRFM, empfiehlt sich der Vergleich zwischen konventionellen und TIRFM-Bildern von ein und derselben Probe. TIRFM ist mit der weit verbreiteten Epifluoreszenzmikroskopie kompatibel und ergänzt so Anwendungen in der Mikroskopie ohne Laser. Sie ist ein leistungsstarkes Verfahren für sämtliche Bereiche in der modernen Zellbiologie, im Gewebe-Engineering sowie im pharmazeutischen Screening und eignet sich insbesondere für Untersuchungen zahlreicher Prozesse an der Zelloberfläche, wie beispielsweise Endo-/Exocytose, Zelladhäsion und die Ausbildung von Verbindungen zwischen Zellen.
Materialien und Methoden Präparation der Zellen
16 Stunden vor dem Imaging wurden menschliche embryonale Nierenzellen (HEK-293) auf 16 mm Deckgläser ausgesät und unter der Verwendung von jetPEI™ (Polyplus- Transfektion) mit YFP (gelb fluoreszierendes Protein)-/ mCherry-markierten Konstrukten transfiziert. Für das Calcium- Imaging wurden die Zellen mit Fura-2 AM (2 µM) beladen, das zunächst in einem extrazellulären Calciumpuffer (121 mM NaCl, 6 mM NaHCO3, 5,4 mM KCl, 5,5 mM D-Glukose, 0,8 mM MgCl2, 25 mM HEPES, 1,8 mM CaCl2, pH 7,4) verdünnt worden war. Dann wurden sie mit einem extrazellulären Calciumpuffer gewaschen und anschließend für eine halbe Stunde stehen gelassen, um die Hydrolyse der lipophilen Sperrgruppen zu ermöglichen. Die Deckgläser wurden dann zum Imaging auf rostfreie Stahlringe montiert und die Zellen in einen extrazellulären Calciumpuffer getaucht. Um die Translokation des STIM1 auszulösen, wurde Thapsigargin (2 µM) in einem Calciumfreien extrazellulären Puffer hinzugefügt, dessen Zusammensetzung der des Calciumpuffers entspricht, mit der Ausnahme, dass hierbei 1 mM EGTA das CaCl2 ersetzt. Durch Hemmung der es Calciumpumpe im endoplasmatischen Retikulum (ER), zeigt das Thapsigargin auf, dass Calcium passiv aus dem ER austritt, wodurch es zu einer erhöhten Konzentration an zytosolischem Calcium kommt. Sekundär aktiviert der Speicherabbau Calciumkanäle in der Plasmamembran, die wiederum einen Calciumzufluss ins Zytosol zulassen.
Imaging
Sämtliche Bilder entstanden mit dem Olympus cell R Imaging-System. Der Aufbau umfasst ein IX81-Mikroskop, ein Apochromat-Objektiv mit 100-fach Vergrößerung und einer numerischen Apertur von 1,45, ein Beleuchtungssystem MT-20, zwei Laser mit den Wellenlängen 488 nm (20 mW) und 561 nm (25 mW) und eine Hamamatsu ORCA ER-Kamera. Für konstante 32 °C sorgte am IX81 eine Klimakammer der Firma Solent Scientific. Bei der Aufnahme der Weitfeld-Epifluoreszenz-Bilder vom YFP und Fura-2 kamen ein 470/40 Anregungsfilter und ein 505 nm Langpassfilter beziehungsweise ein 380/15 nm Anregungsfilter und ein 505 nm Langpassfilter zum Einsatz. Die Aufnahme der TIRFM-Bilder vom YFP und mCherry erfolgte mit 488 beziehungsweise 561 nm Laseranregung sowie einem 527/21 645/24 nm Dual- Bandpass-Emissionsfilter. Gewählt wurden die folgenden Belichtungszeiten: YFP-Weitfeld = 500 ms, YFP-TIRF = 300 ms, mCherry-TIRF = 400 ms, Fura2 = 100 ms. Die kleinsten Taktzeiten (das heißt die minimal benötigte Zeit, um eine Bildreihe aufzunehmen) beliefen sich auf 2007 ms für YFP-Weitfeld + YFP-TIRF sowie 2138 ms für YFP-TIRF + mCherry-TIRF + Fura2- Weitfeld. Für sämtliche Experimente war die Kameraverstärkung auf 100 eingestellt. Die Bildanalyse erfolgte mit der Cell^R-Software von Olympus.
Ergebnisse
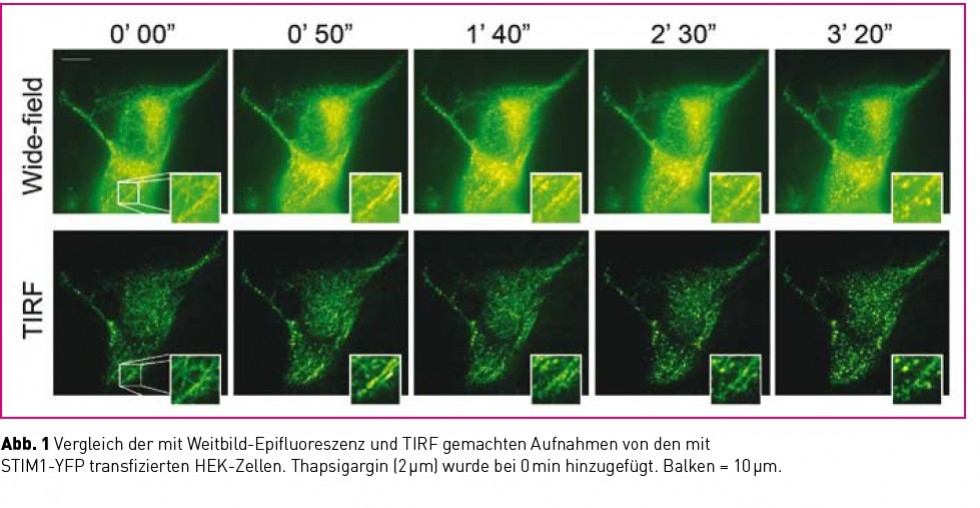
Dank eines hochmodernen Imaging-Systems sowie spezifischer Fluoreszenzsonden war es möglich, TIRFM-Beleuchtung für das Imaging einzusetzen. Dadurch ließen sich die Zellstrukturen an der Plasmamembran, die mit Fluoreszenz-gekennzeichnetem STIM-1 markiert war, ganz einfach und nahezu zeitgleich mit dem intrazellulären Calcium beobachten. Die Abb. 1 vergleicht die Epifluoreszenz und die TIRF-Bilder der mit STIM1-YFP transfizierten HEK-Zellen.
TIRF-Mikroskopie ist die bevorzugte Methode für das hochauflösende Imaging von STIM1- Translokationen/Umverteilung an der Plasmamembran, da keine Unschärfe die Bildqualität beeinträchtigt.
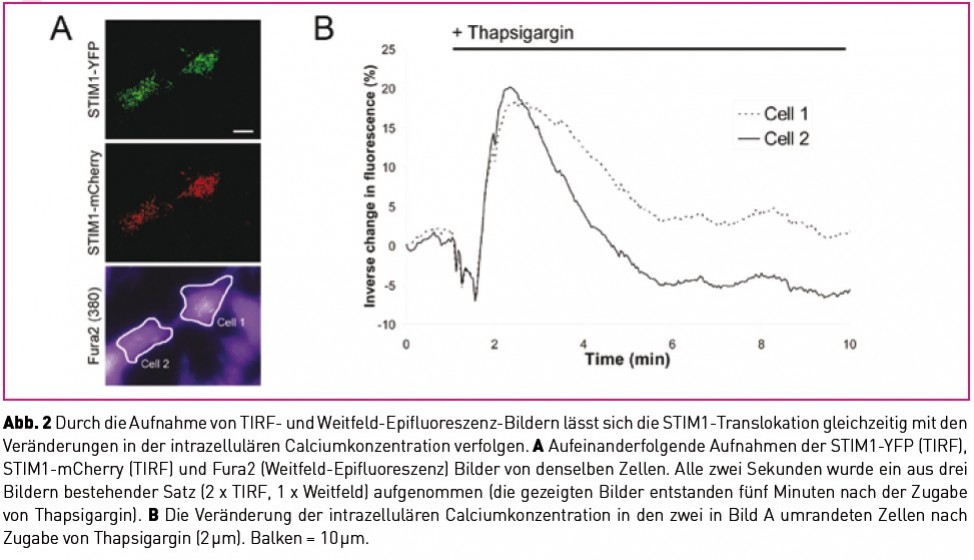
Über die Aufnahme von TIRF-Bildern und Weitfeld- Epifluoreszenz-Bildern (Abb. 2) lässt sich die STIM1-Translokation gleichzeitig mit den Änderungen in der intrazellulären Calciumkonzentration verfolgen.
Abb. 2A zeigt die aufeinanderfolgende Aufnaheme der STIM1-YFP (TIRF), STIM1-mCherry (TIRF) und Fura2 (Weitfeld-Epifluoreszenz) Bilder von denselben Zellen. Alle zwei Sekunden wurde ein aus drei Bildern bestehender Satz (2 x TIRF, 1 x Weitfeld) aufgenommen (die gezeigten Bilder entstanden fünf Minuten nach der Zugabe von Thapsigargin.) Abb. 2B zeigt die Änderung der intrazellulären Calciumkonzentration in den zwei in Bild A umrandeten Zellen nach Zugabe von Thapsigargin (2 µm). Balken = 10 µm.
Perspektiven
Durch die Möglichkeit, das gesamte Potenzial des laserbasierten Fluoreszenz- Imaging auch für die Lebendzell-Mikroskopie auszuschöpfen und das dynamische Imaging von Calcium und STIM1 von ein und derselben Zelle zu kontrollieren, können sich neue Erkenntnisse über die mögliche Rolle der STIM-Proteine in Signaltransduktionsprozessen vom Ca2+- Speicherabbau bis zur Induktion des Ca2+- Zuflusses ergeben. Darüber hinaus lässt sich mit dieser Technologie auch die Eigenschaft der STIM-Proteine als CA2+-Sensoren genauer untersuchen und ebenso, wie sich dieser Prozess auf die örtlichen Regionen nahe der Plasmamembran und dem ER auswirken könnte.
Fotos: © Simon Walker
Literatur beim Autor
Danksagungen Unser besonderer Dank gilt R. S. Lewis (Stanford) für die Bereitstellung des mCherry-STIM1 und S. Muallem (UTSW, Texas) für das YFP-STIM1.
Stichwörter:
TIRF-Mikroskopie
|
L&M 2 / 2009
Diese Artikel wurden veröffentlicht in Ausgabe L&M 2 / 2009.
Das komplette Heft zum kostenlosen Download finden Sie hier:
zum Download
Der Autor:
Weitere Artikel online lesen
News
Mit dem HPLC-Säulenkonfigurator unter www.analytics-shop.com können Sie stets die passende Säule für jedes Trennproblem finden. Dank innovativer Filtermöglichkeiten können Sie in Sekundenschnelle nach gewünschtem Durchmesser, Länge, Porengröße, Säulenbezeichnung u.v.m. selektieren. So erhalten Sie aus über 70.000 verschiedenen HPLC-Säulen das passende Ergebnis für Ihre Anwendung und können zwischen allen gängigen Herstellern wie Agilent, Waters, ThermoScientific, Merck, Sigma-Aldrich, Chiral, Macherey-Nagel u.v.a. wählen. Ergänzend stehen Ihnen die HPLC-Experten von Altmann Analytik beratend zur Seite – testen Sie jetzt den kostenlosen HPLC-Säulenkonfigurator!
© Text und Bild: Altmann Analytik
Aufnahme, Dokumentation und Teilen von Ergebnissen mit ZEISS Stemi 305 und ZEISS Stemi 508
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen.
© Text und Bild: Carl Zeiss Microscopy GmbH
|