|
Forscher
>
Prof. Dr. Jürgen Bajorath
>
Neue Visualisierungskonzepte für die medizinische Chemie
Neue Visualisierungskonzepte für die medizinische ChemieWanderungen durch AktivitätslandschaftenMassives Wachstum biologischer und chemischer Daten unterstützt wissensbasierte Arbeitsweisen in der Pharmaforschung, stellt allerdings auch hohe Anforderungen an Datenanalytik und experimentelles Design. Das betrifft auch die medizinische Chemie. Konventionelle Ansätze für die Analyse von Strukturfunktionsbeziehungen niedermole-kularer Verbindungen (Structure-Activity Relationship; SAR) sind nicht mehr ausreichend, um stetig wachsende Datenmengen (Moleküle und deren In-vitro-Aktivitäten) auszuwerten und experimentell überprüfbare Hypothesen zu generieren. In den letzten Jahren sind neue computergestützte Methoden für die systematische SAR-Analyse großer Datensätze entwickelt worden, die einen besonderen Schwerpunkt auf Visualisierung setzen, um komplexe SAR-Muster intuitiv erfassbar zu machen und die Identifizierung von Schlüsselverbindungen zu unterstützen. Subjektive Kriterien in der Chemie 
Chemiker werden überwiegend mithilfe zweidimensionaler molekularer Repräsentationen ausgebildet (so genannte molekulare Graphen). In der medizinischen Chemie spielt die Analyse von SARs eine zentrale Rolle im Rahmen der chemischen Optimierung von Leitstrukturen. Die SAR-Analyse wird traditionell auf der Basis molekularer Graphen durchgeführt. Dabei werden SAR-Daten in Substituenten-Tabellen (R-Group Table; RGT) organisiert, die die Kernstruktur einer Verbindungsreihe, alle vorkommenden Substituenten und die biologischen Aktivitäten der korrespondierenden Moleküle auflisten. Diese RGTs sind bis heute ein konventionelles Rüstzeug des medizinischen Chemikers. Traditionell konzentriert sich die chemische Optimierung auf individuelle Verbindungsreihen. Man arbeitet an einer bestimmten Kernstruktur, stellt analoge Verbindungen mit unterschiedlichen Substituenten her, bestimmt deren biologische Aktivität und registriert die Ergebnisse in RGTs. Falls der Fortschritt mit einer Verbindungsreihe in der Leitstrukturoptimierung nicht ausreichend ist, wird eine andere Serie bearbeitet. Dabei wird natürlich nicht nur die primäre Wirksamkeit eines Moleküls erhöht, sondern es müssen mehrere optimierungsrelevante Parameter ins Kalkül gezogen werden (z.B. Löslichkeit oder metabolische Stabilität). RGTs sind eine sinnvolle Datenstruktur, solange man an individuellen Verbindungsreihen arbeitet und die Zahl der Analoge begrenzt ist. So ist es kaum möglich, hunderte von Analogen unterschiedlicher Serien mit dem Auge des Chemikers einer subjektiven Analyse zu unterziehen und SAR-Regeln abzuleiten, die zu einer sinnvollen Optimierung führen. Hier stoßen wir rasch an unsere Grenzen. Dennoch spielen subjektive Kriterien, chemische Intuition und individuelle Erfahrung eine Schlüsselrolle in der medizinischen Chemie – und der Erfolg gibt chemischem Scharfsinn oft Recht. Wir wissen aber auch, dass selbst erfahrene medizinische Chemiker selten in ihrer Analyse übereinstimmen, welche Moleküle bevorzugte Leitstrukturen oder Medikamentkandidaten darstellen [1]. Zusätzlich ist unsere Perzeption chemischer Strukturen und Eigenschaften kontextabhängig. So hängen unsere Schlussfolgerungen typischerweise von der Reihenfolge ab, in der uns Moleküle präsentiert werden [1]. Trotz der vielen Erfolge subjektiver Analyse und chemischer Intuition ist in der medizinischen Chemie noch viel Raum für „objektive“ Datenanalytik und wissensbasiertes molekulares Design. Datenflut In den letzten Jahren erleben wir ein fast exponentielles Wachstum von SAR-Daten und ein Ende ist nicht in Sicht. Diese Datenflut führt nicht nur zu einer noch nie da gewesenen Wissensbasis, die allerdings erst einmal erschlossen werden muss, sondern erschwert auch traditionelle Arbeitsweisen in der medizinischen Chemie. SAR-Daten nehmen innerhalb der Pharmaindustrie und auch im öffentlichen Sektor mit großer Geschwindigkeit zu. So sind in großen öffentlichen Datenbanken wie ChEMBL [2] oder PubChem [3] zurzeit bereits mehr als 10 Mio. aktive Moleküle verfügbar, die zu einem großen Teil mit mehreren Zielproteinen assoziiert sind. Für attraktive therapeutische Zielproteine sind in aller Regel bereits viele Verbindungsreihen mit oft sehr unterschiedlichem SAR-Informationsgehalt vorhanden. Zusätzlich zu den großen Datenvolumen erschwert die Heterogenität der SAR- Informationen eine sinnvolle und erfolgversprechende Auswertung und Umsetzung in zunehmend wirksamere Kandidatenmoleküle. Lernen von großen Mengen heterogener SAR-Daten ist in der Tat zu einer großen Herausforderung in der medizinischen Chemie geworden. Computergestützte Analyse und Aktivitätsvorhersagen Neue Anforderungen an die Datenanalyse gehen ganz eindeutig über die Kapazität subjektiver Vorgehensweisen mithilfe von RGTs hinaus. Sollte man deshalb nicht erwarten, dass Computermethoden einen systematischeren und objektiveren Zugang zu diesen Daten und deren Umsetzung ermöglichen? Ganz ohne Zweifel. Allerdings hat computergestütztes Arbeiten in der medizinischen Chemie bisher eine im Wesentlichen andere Rolle gespielt. Bei einer Diskussion von Computermethoden mit Relevanz für die medizinische Chemie sollte man unterscheiden zwischen Methoden des modernen „Drug Designs“, die in aller Regel von Computerchemikern entwickelt und angewendet werden, oft relativ weit entfernt von der Praxis der medizinischen Chemie, und Methoden, die seit langer Zeit praxisnah angewendet werden. Hierbei handelt es sich in erster Linie um Methoden, die auf dem mittlerweile klassischen „Quantitativen SAR“ (QSAR)-Paradigma basieren [4]. Der ursprünglichen Konzeption folgend, versucht QSAR lineare mathematische Modelle biologischer Aktivität auf der Basis von molekularen Deskriptoren zu entwickeln. QSAR-Modelle werden für individuelle Verbindungsreihen mit Molekülen bekannter Aktivität generiert und dann angewendet, um die Wirksamkeit neuer Analoge vorherzusagen. Obwohl gängige QSAR-Methoden oft in ihren Details variieren, versuchen alle QSAR-Analysen im Grunde die Kernfrage zu adressieren, die für jeden praktizierenden medizinischen Chemiker im Mittelpunkt seiner Arbeit steht: Welche Verbindung soll ich als nächste synthetisieren? Dieser Zusammenhang erklärt die Popularität der praxisnahen QSAR-Analyse in der medizinischen Chemie und erklärt auch, warum medizinische Chemiker in aller Regel mehr an Aktivitätsvorhersagen interessiert sind als an computergestützter SAR-Datensuche und -analyse. Bei der derzeitigen Datenexplosion stellt diese Orientierung natürlich auch ein Problem für den Fortschritt der medizinischen Chemie dar, das inzwischen zunehmend erkannt und diskutiert wird. Neue Konzepte Die Erkenntnis, dass Computeranwendungen in der medizinischen Chemie über konventionelle (Q)SAR-Analysen hinausgehen sollten, um vermehrte Wissensgewinnung aus großen Mengen interner und externer SAR-Daten zu ermöglichen, hat in der letzten Zeit die Entwicklung konzeptionell neuer Methoden katalysiert. In schwierigen Zeiten für die Pharmaindustrie kann man es sich natürlich nicht leisten, auf dieses Wissen zu verzichten. Man muß aus vielen mehr oder weniger abgeschlossenen (erfolgreichen oder erfolglosen) Projekten vermehrt lernen, um datenorientierte Planungen und Entscheidungen zu ermöglichen. So sind in den letzten Jahren neue Computermethoden zur groß angelegten und systematischen Analyse heterogener SAR-Daten entwickelt worden, die z. B. in der Lage sind, die Evolution von SARs über viele unterschiedliche Molekülserien zu verfolgen, sowohl komplexe als auch subtile SAR Trends zu detektieren und Verbindungen mit besonders hohem SAR-Informationsgehalt zu identifizieren. Zum Beispiel werden neue numerische SAR-Analysefunktionen dazu verwendet, molekulare Struktur- und Aktivitätsähnlichkeiten systematisch zu vergleichen und SAR-Informationsgehalt konsistent zu quantifizieren [5]. Zusätzlich sind effiziente Algorithmen entwickelt worden, um strukturverwandte Molekülpaare mit genau definierten Strukturunterschieden aus großen Datensätzen zu extrahieren [6] und mit SAR-Information zu assoziieren [7]. Darüber hinaus spielen Visualisierungsmethoden eine bedeutende Rolle [8]. SAR-Visualisierung
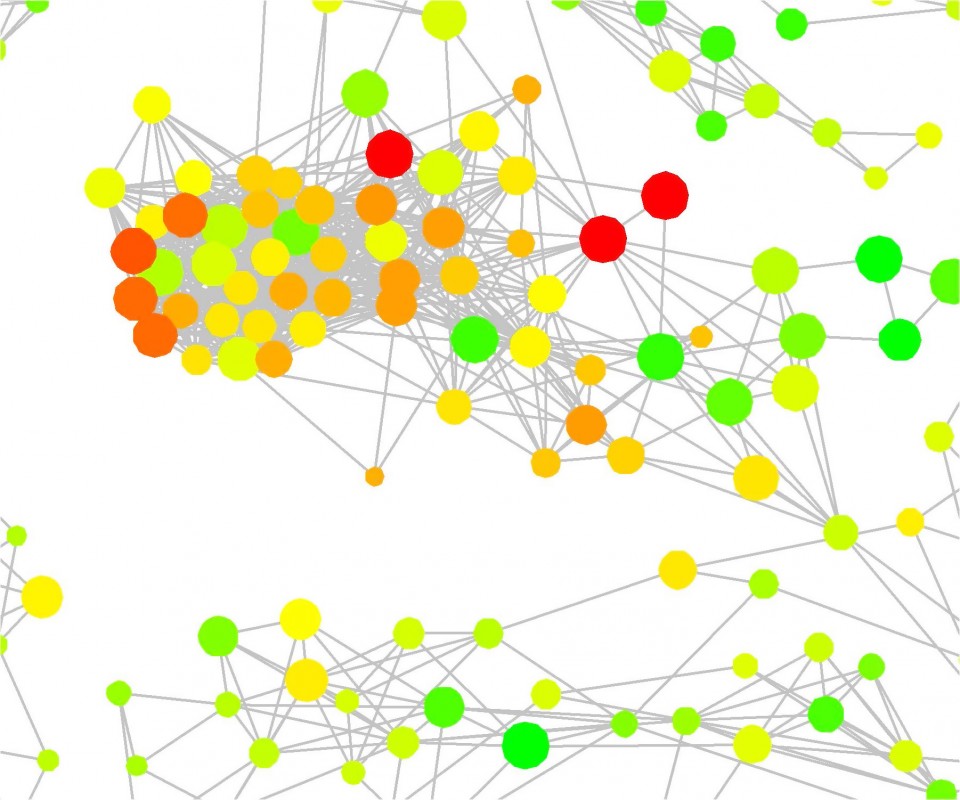
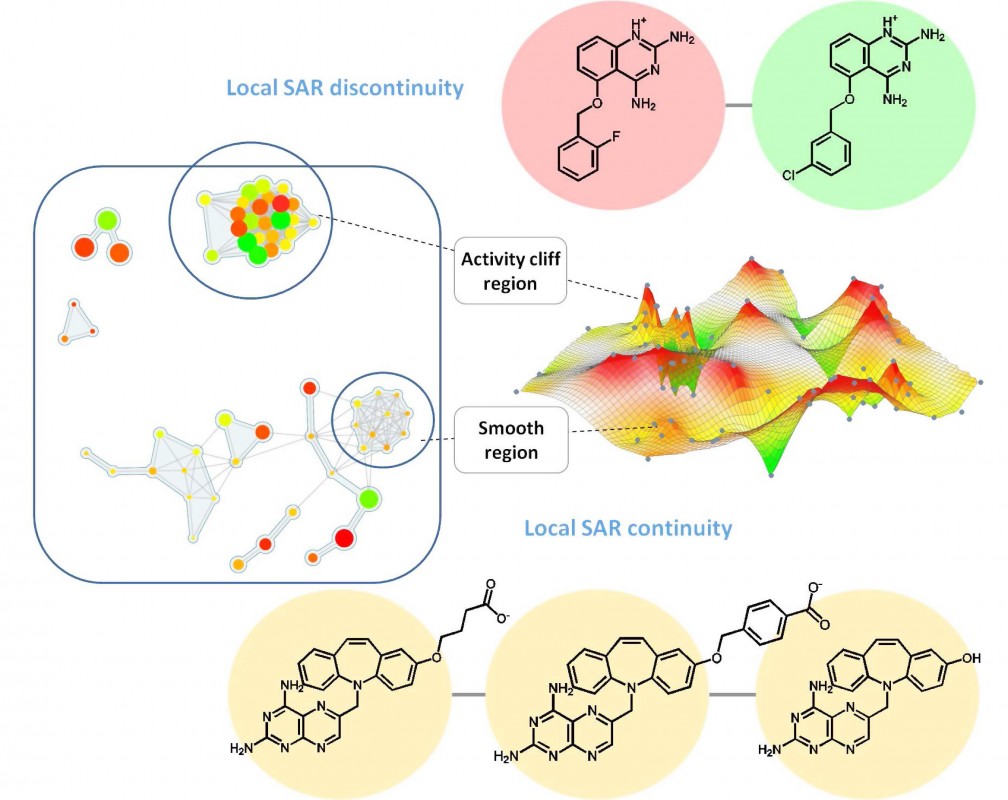
Die Anwendbarkeit numerischer Beschreibungen von SAR-Eigenschaften ist in der praktischen medizinischen Chemie oft begrenzt. Die Ergebnisse groß angelegter SAR-Analysen müssen Chemikern in intuitiver Weise zugänglich gemacht werden, gerade dann, wenn SAR-Muster komplex sind. Für große Datensätze sind dazu neue Visualisierungsmethoden erforderlich, die zunehmend an Popularität gewinnen.
Literatur
Foto: © istockphoto.com| |
L&M 3 / 2013
Das komplette Heft zum kostenlosen Download finden Sie hier: zum Download Der Autor:NewsSchnell und einfach die passende Trennsäule finden
© Text und Bild: Altmann Analytik ZEISS stellt neue Stereomikroskope vor
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen. © Text und Bild: Carl Zeiss Microscopy GmbH |