


|
Forscher
>
Prof. Dr. Albert Jeltsch
>
Die Entschlüsselung des epigenetischen Codes
Die Entschlüsselung des epigenetischen CodesEinblicke in die Betriebsanleitung des menschlichen Genoms
Das menschliche Genom umfasst 3 Mrd. Buchstaben in Form von Basenpaaren in der DNA-Sequenz. Es codiert für etwa 20.000 bis 25.000 Gene, die üblicherweise in verschiedenen Varianten abgelesen werden können, und zusätzlich noch für eine große Anzahl von funktionellen RNA-Molekülen, die nicht in Proteine umgesetzt werden. Diese große Menge an Information enthält alle erforderlichen Anweisungen zum Erzeugen der verschiedenen menschlichen Zelltypen in jeder möglichen Entwicklungsstufe und unter beliebigen Umweltbedingungen. Jede Zelle verwendet aber jeweils nur einen kleinen Bruchteil dieser gewaltigen Datenmenge. Die Regelung des Zugangs zu genetischen Informationen erfolgt durch sogenannte epigenetische Mechanismen. Diese geben nicht nur das für den Zelltyp spezifische Genexpressionsmuster vor, sondern auch das für den Zelltyp spezifische mRNA-Splicing und die Erzeugung der zahlreichen, nicht codierenden RNAs, die regulierende Rollen haben. Im Allgemeinen ist die epigenetische Regelung mit zwei Herausforderungen konfrontiert. Einerseits muss sie stabil sein, um zelluläre Identitäten zu erhalten, und andererseits muss sie flexibel sein, um Entwicklungsprozesse und ein Reagieren auf Umweltsignale zu ermöglichen. In der frühen Evolution war die Entwicklung von leistungsfähigen epigenetischen Systemen ein kritischer Faktor im Ursprung von mehrzelligen Organismen [1]. Heute treten Krankheiten auf, wenn epigenetische Systeme versagen [2, 3]. Krebs ist beispielsweise die Folge der Dedifferenzierung von Zellen in einen sich schnell teilenden Zustand. Und Diabetes als ein Beispiel für eine Stoffwechselkrankheit tritt auf, weil die Reaktion auf Insulinsignale nicht mehr funktioniert. Wie sieht epigenetische Information aus? Die 2 m lange menschliche DNA ist an Histonproteine gebunden und extrem kondensiert, um in den Zellkern mit einem Durchmesser in der Größenordnung von 10-6 m zu passen. Auf der ersten Ebene sind 147 Basenpaare von DNA um ein Histonoktamer gewickelt, das zwei Kopien der Histone H2A, H2B, H3 und H4 umfasst, die mit der DNA ein sogenanntes Nukleosom bilden. Epigenetische Information ist in kovalenten Modifikationen auf der DNA und den Histonproteinen verschlüsselt. Auf der DNA sind die Cytosinbasen methyliert (vor allem in einem CpG-Kontext) und das Methylcytosin kann weiter zu Hydroxymethylcytosin und in höheren Oxidationsformen umgewandelt sein. Auf den Histonen treten Modifikationen vor Abb. 2 Modell des DNA-Methyltransferase-DNMT3A/3L-Komplexes (grün), der an die Linker-DNA neben einem Nukleosom gebunden ist. Die ADD- (rot) und PWWP-Domänen (orangefarben) von DNMT3A sind nur für eine der Untereinheiten dargestellt und binden an das H3-N-Ende an Lysin 4 und Lysin 36. allem an den verlängerten N-Enden auf, die aus dem globulären Nukleosom herausragen. Modifikationen umfassen vor allem Methylierungen von Lysin und Arginin, Acetylierung von Lysin, Phosphorylierung von Serin und Threonin und Ubiquitinierung von Lysin [4]. Über wie viel epigenetische Information verfügen wir? Angenommen, etwa 3 Prozent der 850 Mio. Cytosinreste im haploiden Genom sind methyliert, so entspricht dies etwa 540 MB Information. Histonmodifikationen treten vor allem an den H3- und H4-Enden auf, die in jeweils zwei Kopien pro Nukleosom vorhanden sind. Angenommen, etwa 50 verschiedene Modifizierungen sind vorhanden, so beträgt der Informationsgehalt der etwa 16 Mio. Nukleosomen etwa 190 MB, sodass die gesamte epigenetische Information in einer einzelnen Zelle etwa 730 MB entspricht, etwa der Hälfte des Informationsgehalts des gesamten menschlichen Genoms (1,5 GB). Da der menschliche Körper aber aus etwa 200 Zelltypen besteht, die sich in ihrem epigenetischem Zustand unterscheiden, ist die gesamte epigenetische Information eines Menschen viel größer als die im Genom verschlüsselte Information, was darauf hinweist, dass es wesentlich komplexer und schwieriger ist, den Zugang zur Genominformation zu kontrollieren, als die Daten selbst zu speichern.
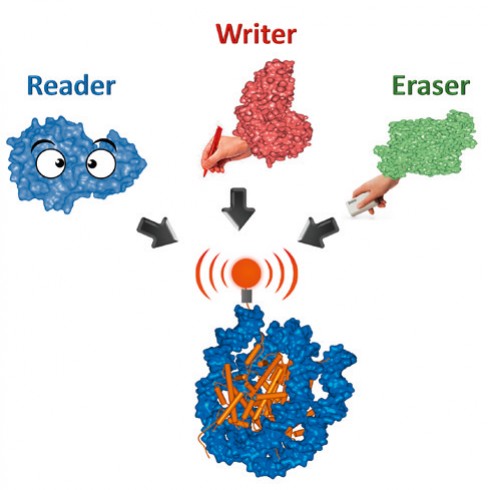
Abb.1 Epigenetische Signale an Nukleosomen werden von Schreibenzymen („Writer“) gesetzt, von Löschenzymen („Eraser“) entfernt und von Lesedomänen („Reader“) abgelesen.
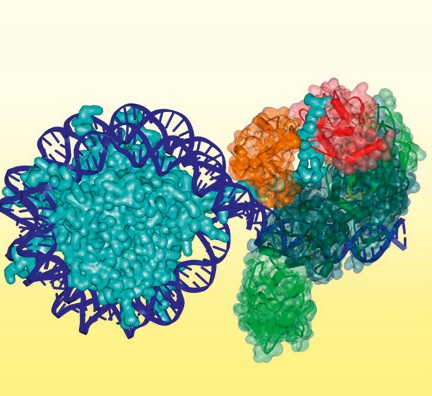
Abb.2 Modell des DNA-Methyltransferase-DNMT3A/3L-Komplexes (grün), der an die Linker-DNA neben einem Nukleosom gebunden ist. Die ADD- (rot) und PWWP-Domänen (orangefarben) von DNMT3A sind nur für eine der Untereinheiten dargestellt und binden an das H3-N-Ende an Lysin 4 und Lysin 36.
Inwiefern unterscheidet sich die epigenetische Regelung von anderen Regelungsarten? Der wesentliche Unterschied zwischen epigenetischer Regulation und anderen Regulationstypen in der Zelle besteht darin, dass die epigenetische Regulation stabiler ist und durch Zellteilungen und selbst durch die Keimbahn vererbt werden kann, obwohl die Information durch die DNA-Vervielfältigung im Zuge der Synthese eines neuen DNA-Strangs und das Hinzufügen neu synthetisierter Histonproteine verdünnt wird. Dies kann durch Verwenden von epigenetischen Kopierprozessen erzielt werden, die bestehende epigenetische Information identifizieren und diese auf die neu hinzugefügten Komponenten kopieren [5]. Die so gebildeten positiven Feedback-Schleifen sind ein wesentliches Merkmal epigenetischer Vorgänge. Auf dieser Grundlage lautet eine übliche Definition von epigenetischen Prozessen, dass sie vererbbare Information zum Genom hinzufügen, die aber grundsätzlich reversibel ist. Diese Definition ist zwar leistungsfähig, aber unvorteilhafterweise mit dem Konzept der Zellteilung verknüpft. Somit könnte epigenetische Information in einer sich nicht teilenden Zelle wie einem Neuron nicht existieren. Es ist nun aber wenig sinnvoll, einen molekularen Prozess wie die DNA-Methylierung in einem Zelltyp als „epigenetisch“ zu bezeichnen und in einem anderen nicht. Daher wird der Begriff „epigenetisch“ im weiteren Sinne für alle molekularen Prozesse verwendet, die eine epigenetische Rolle in einigen Zelltypen oder Modellsystemen spielen. So wird dieser Begriff auch hier verwendet. Wie funktionieren epigenetische Signale? Im Allgemeinen umfasst das epigenetische System Enzyme, die epigenetische Marker setzen oder entfernen, sogenannte Schreiber und Löscher, und Proteine, die sich spezifisch an DNA oder Histone binden, wenn diese eine definierte Modifikation tragen, sogenannte Leser. Das epigenetische Muster wird durch eine Rekrutierung von Schreibern und Löschern an bestimmte Stellen im Genom und die Regelung ihrer Aktivitäten gebildet. Biologische Wirkungen werden durch Leser vermittelt, die andere Chromatinfaktoren an genomische Regionen bringen, die bestimmte Modifizierungen tragen, und dadurch verschiedene biologische Folgen wie beispielsweise die Kondensation des Chromatins und Bildung von Heterochromatin, die Genexpression oder die DNA-Reparatur auslösen. Leser sind ebenfalls an der Adressierung der Schreiber und Löscher beteiligt, sodass das Einfügen und Entfernen eines epigenetischen Markers vom Vorhandensein und/ oder Fehlen anderer Marker abhängt, was komplexe und äußerst wichtige Feedback-Zyklen bildet. Diese sind besonders wichtig für die Aufrechterhaltung von epigenetischen Mustern in der Form von DNA-Methylierung und Histonmodifikationen, wobei verschiedene und noch nicht vollständig erforschte Feedback-Schleifen existieren, die ein stabiles Vererben dieser Modifizierungen gewährleisten [5].

Abb.3 Struktur des katalytischen Bereichs der MLL3-Histon-Methyltransferase. Das Substratpeptid ist in Orange dargestellt, der Kofaktor AdoMet in Gelb. Zwei Aminosäurenreste, die häufig bei Krebs mutiert sind, sind in Rot und Grün dargestellt.
Gibt es Anwendungen für unser Wissen in der molekularen Epigenetik? Aufgrund der wesentlichen Rolle von Fehlern in der epigenetischen Information für Gesundheit und Krankheit sowie der Reversibilität von epigenetischen Signalen ist die Epigenom-Editierung, die lokusspezifische Bearbeitung von epigenetischer Information, eine vielversprechende neuartige Technologie [7]. Sie kann durch das Verknüpfen eines epigenetischen Enzyms mit einem DNA-bindenden Proteinfaktor erzielt werden. Nach der Expression in der Zelle bringt der DNA-bindende Teil das Enzym zum Zielort, wo die epigenetische Information anschließend bearbeitet und verändert wird. Hierfür gibt es zahlreiche Anwendungen, etwa die Abschaltung von Onkogenen oder Virusrezeptoren oder das Anschalten von Tumorsuppressorgenen. Darüber hinaus könnten fehlerhafte Regulationswege bei Stoffwechselkrankheiten oder im Gehirn korrigiert werden. Eine epigenetische Bearbeitung könnte weiterhin ein Weg zur rationalen Neuprogrammierung von Zellen in der Erzeugung von patienteneigenen iPS-Zellen und ihrer Entwicklung zu differenzierten Zellen sein, die dann therapeutisch verwendbar sind. Unsere Forschungsschwerpunkte In unserem Labor untersuchen wir, wie epigenetische Muster in Chromatin erzeugt und erhalten werden, wie sie zum Auslösen von biologischen Reaktionen gelesen werden und wie epigenetische Information bearbeitet werden kann. Um das Setzen und Erhalten von epigenetischen Markern zu verstehen, wird der Mechanismus von zwei Gruppen von epigenetischen Schreibern (DNA-Methyltransferasen und Histonprotein-Methyltransferasen) auf molekularer Ebene untersucht. Wir untersuchen die katalytischen Mechanismen und Aktivitäten dieser Enzyme in vitro in Reaktionsgefäßen, aber auch in lebenden Zellen und studieren, wie die Lokalisierung und Regulation dieser Enzyme erfolgt [7, 8]. Dies beinhaltet die Regulation der Enzymaktivität durch posttranslationale Modifikationen der Enzyme sowie ihre allosterische Regulation als Reaktion auf das Binden von Komplexpartnern oder spezifischen epigenetischen Markern. Unsere Untersuchungen führten zu der Erkenntnis, dass viele Histonprotein- Lysin-Methyltransferasen zusätzliche Signalfunktionen in der Methylierung von anderen Proteinen haben [9, 10]. Ferner untersuchen wir, wie Lesedomänen mit hypermodifizierten Histonpeptidenden interagieren und wie sich verschiedene Modifikationen gegenseitig beeinflussen. Ein Schwerpunkt dieser Arbeiten besteht im Verstehen, wie kombinatorische Muster von Modifikationen analysiert werden können und wie sie verschiedene biologische Ergebnisse vermitteln. Schließlich entwickeln wir Strategien und Verfahren für die Epigenom-Editierung und verwenden diese als Ansatz zum Untersuchen der komplexen Architektur des Chromatinmodifikationsnetzes durch zielgerichtete Veränderung einzelner Modifikationen.
-> albert.jeltsch@ibc.uni-stuttgart.de
|
L&M 4 / 2016
Das komplette Heft zum kostenlosen Download finden Sie hier: zum Download Der Autor:NewsSchnell und einfach die passende Trennsäule finden
© Text und Bild: Altmann Analytik ZEISS stellt neue Stereomikroskope vor
ZEISS stellt zwei neue kompakte Greenough-Stereomikroskope für Ausbildung, Laborroutine und industrielle Inspektion vor: ZEISS Stemi 305 und ZEISS Stemi 508. Anwender sehen ihre Proben farbig, dreidimensional, kontrastreich sowie frei von Verzerrungen oder Farbsäumen. © Text und Bild: Carl Zeiss Microscopy GmbH |

